COR Corona COVID-19 forum
Alles over het corona / COVID-19 virus.





Nextstrain is een open-source project waar onze overheid ook aan meewerkt.
Van de afgenomen samples, wordt informatie gedeeld vanuit de hele wereld en met elkaar vergeleken. Omdat het RNA van het virus net zo opgebouwd is als het DNA van de mens, is het mogelijk om a.d.v. de mutaties te bekijken of er een relatie is tussen twee verschillende samples en hoe lang het geleden is dat deze van elkaar gescheiden zijn.
Lees hier meer over virussen op wiki, en hier over het mappen (ENG) van genomen.
Al deze gegevens worden aan elkaar gekoppeld, waarmee het verloop van het virus in kaart is te brengen.
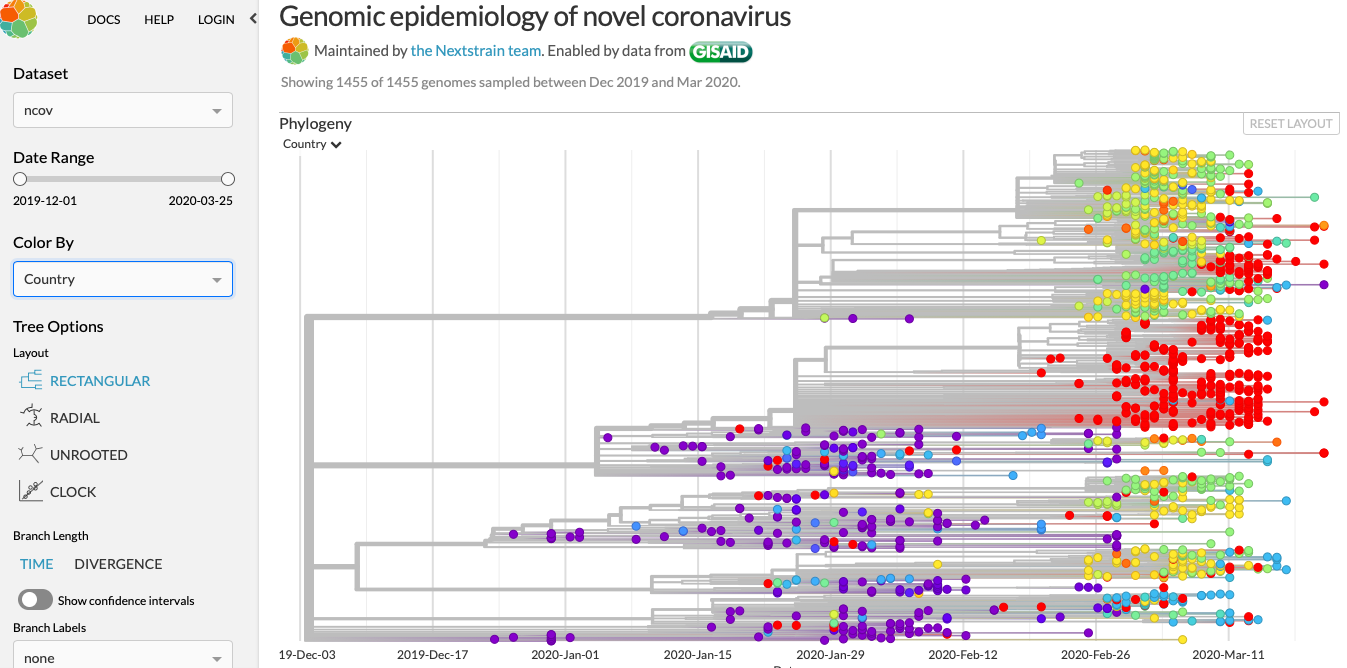
Per land is te zien hoe het verloop in tijd is van de verschillende mutaties van het virus
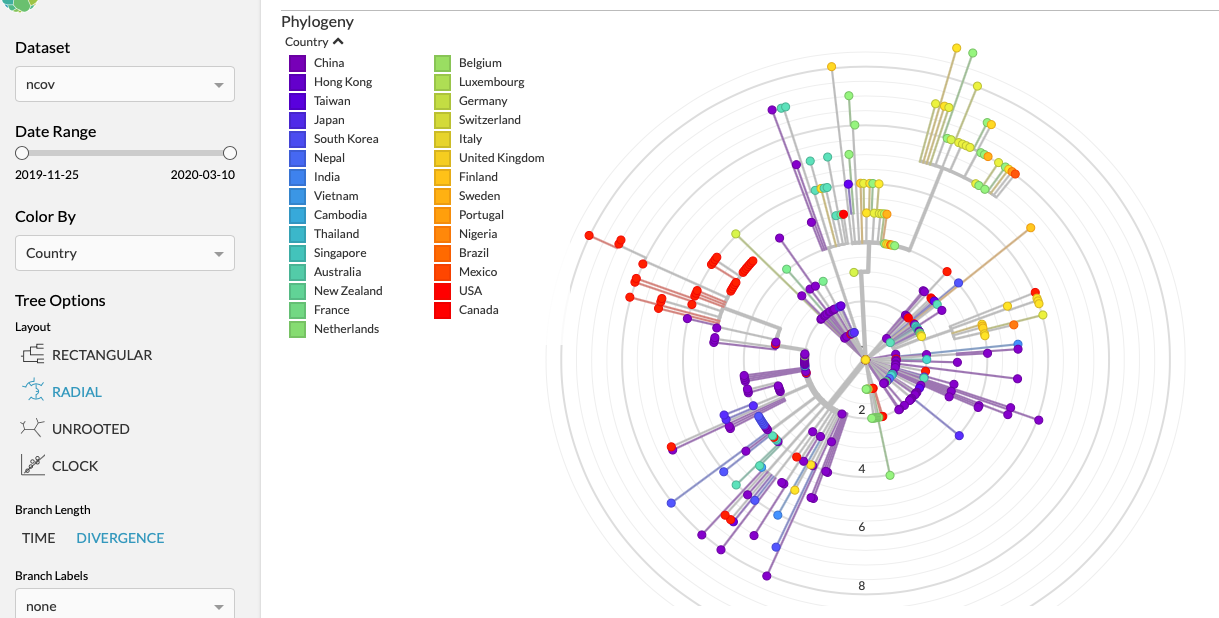
Onderin is het ook mogelijk om filters in te stellen per land of regio, waarmee duidelijk te zien is dat de US/Canada niet zijn besmet via ons, of andersom.
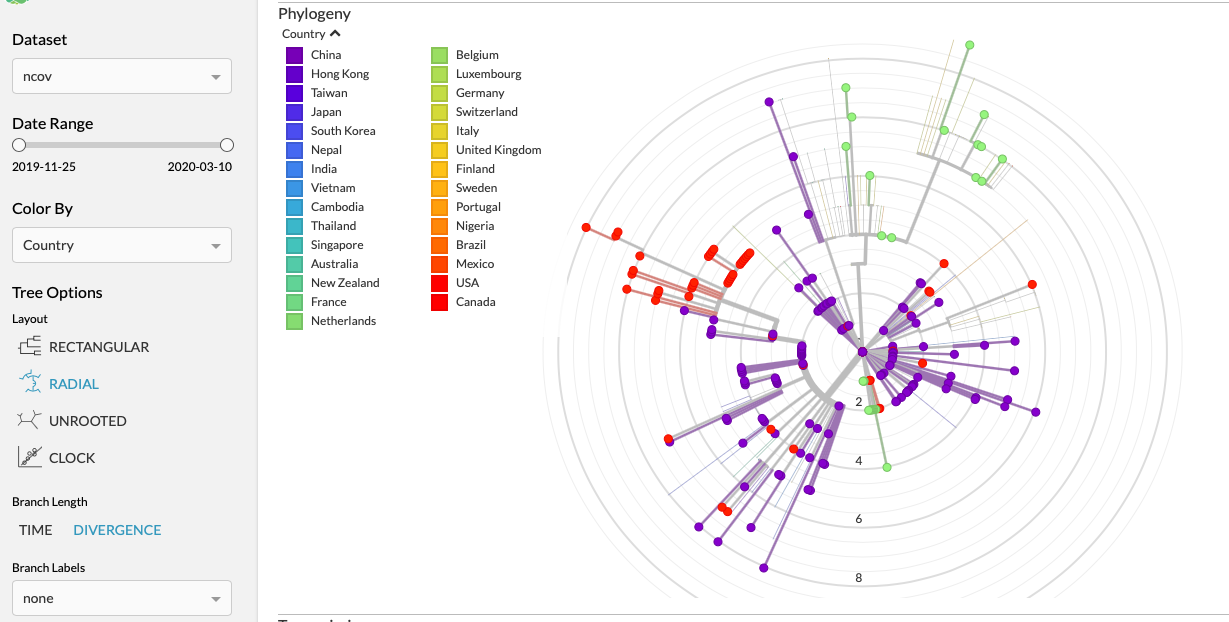
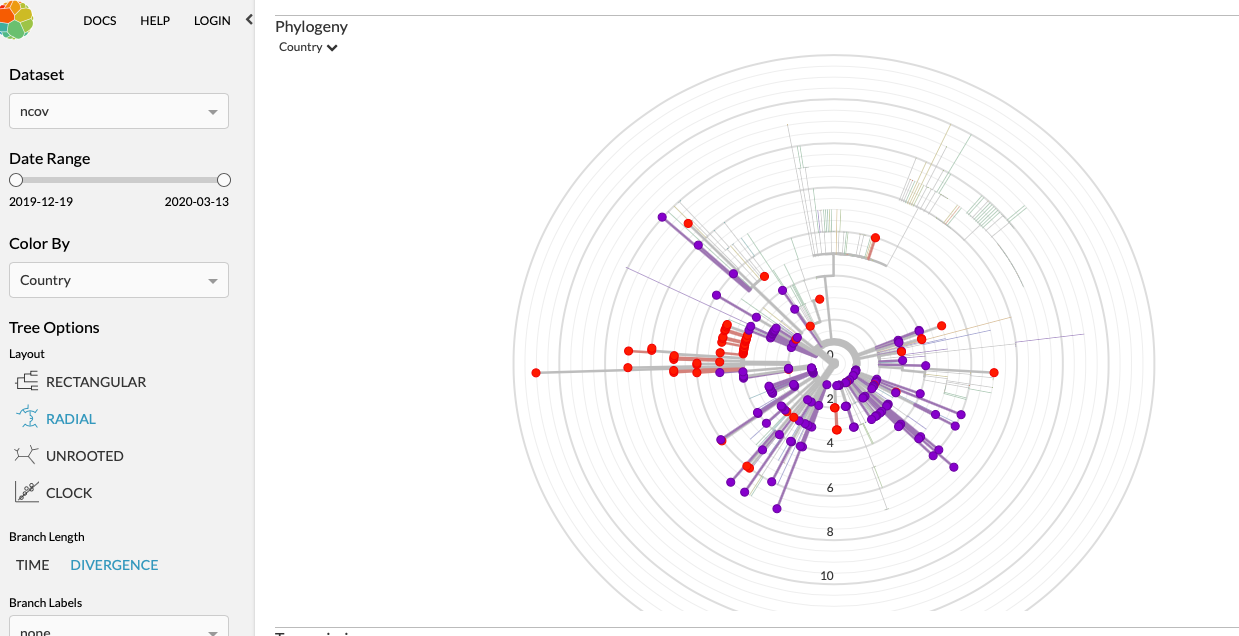
Hier is duidelijk te zien dat er twee verschillende 'strains' zijn ontstaan; het S en L type. Ondanks eerdere berichten dat er een "agressieve" soort zou zijn, is het te vroeg om te zeggen of dit zo is. Men mag altijd hopen dat wij een milde vorm zullen treffen, maar dat zou betekenen dat er ergens anders een zwarte vorm heerst. Brrr!
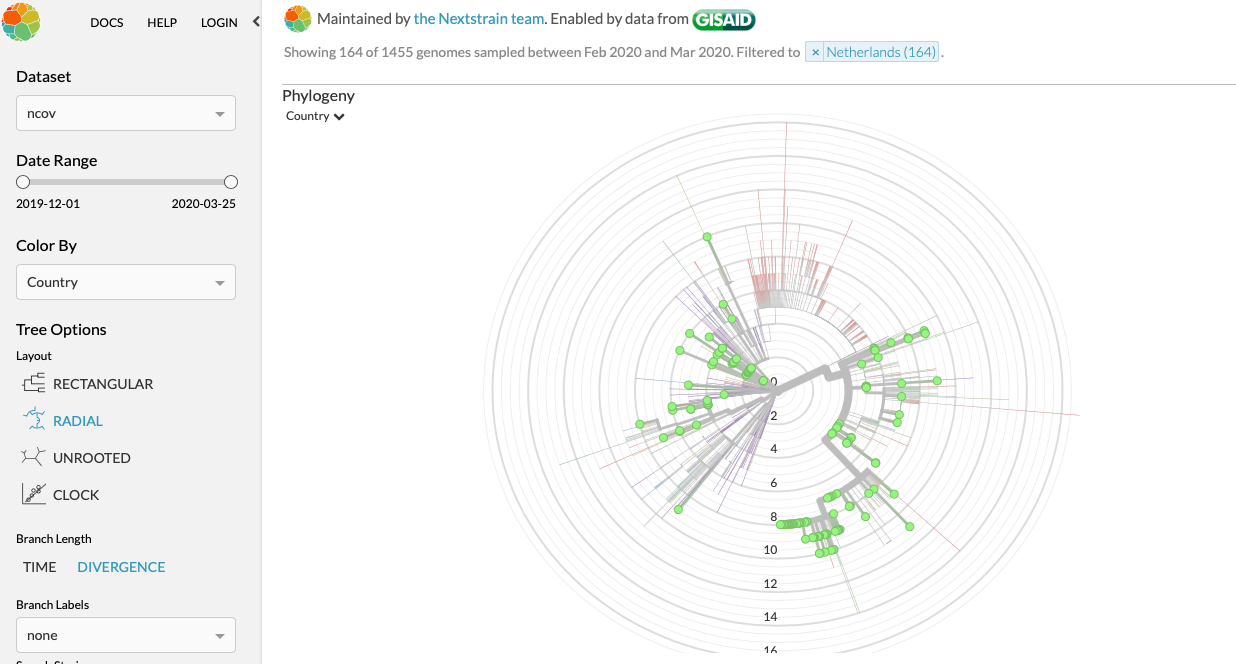
Alleen een filter op NL laat zien dat onze besmettingen uit twee vertakkingen komen;

de bovenste
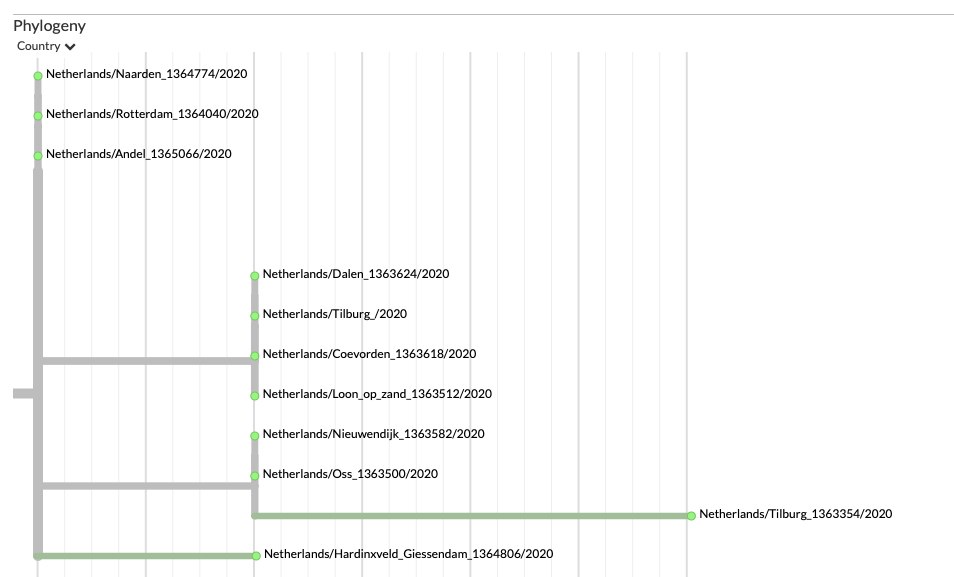
en de onderste
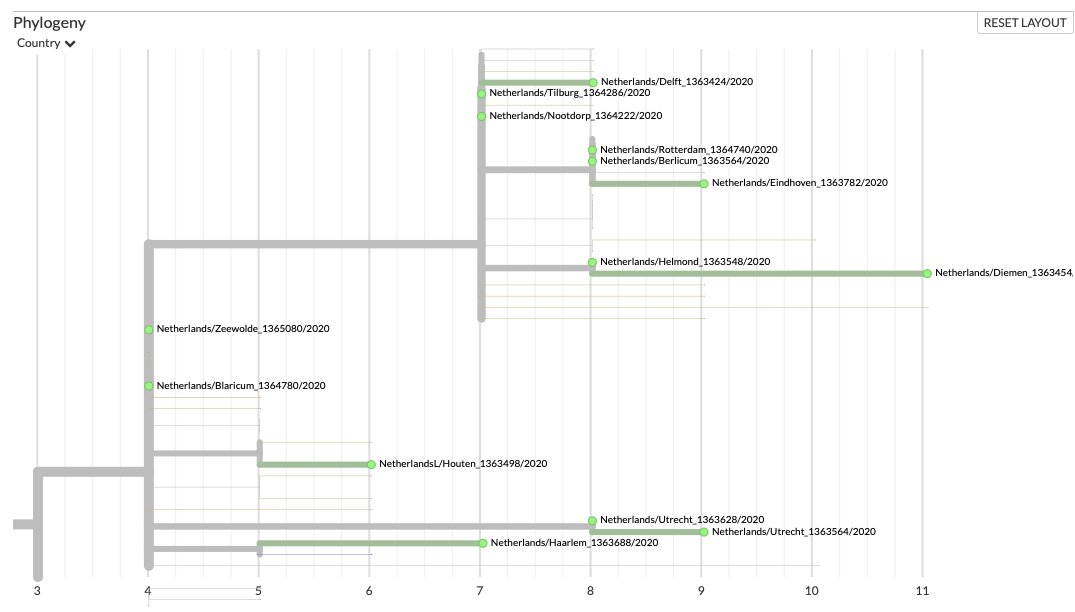
Ons eerste gemeten slachtoffer

Bron: Nextstrain

Interessant project.
Het nut van trump z'n populistische maatregel wordt ook meteen duidelijk 0,0 dus.
0,0 dus.
Het nut van trump z'n populistische maatregel wordt ook meteen duidelijk

Ben geen bioloog, maar geloof niet dat je dat kan stellen.quote:Op donderdag 12 maart 2020 14:12 schreef THEFXR het volgende:
hoe meer vertakking hoe zwakker het virus?
De meeste mutaties vinden plaats in het niet-coderende gedeelte van het RNA en hebben dus geen gevolg voor het virus zelf. Bij een mutatie aan een coderend genoom kan het zijn dat de eigenschappen veranderen; Snellere verspreiding, afwijkende incubatietijd of heftigere/mildere gevolgen.
Ik vind dit een super interessant topic, hulde voor het aanmaken ervan!
Nu een paar vragen, want ik weet weinig van biologie.
Wat kunnen we met deze informatie? Is er bijvoorbeeld uit te destilleren wat de eigenschappen zijn van de takken die in Nederland actief zijn?
Kan bijvoorbeeld herleid worden waar vandaan de Brabantse nog niet teruggeleide besmettingen komen?
Nu een paar vragen, want ik weet weinig van biologie.
Wat kunnen we met deze informatie? Is er bijvoorbeeld uit te destilleren wat de eigenschappen zijn van de takken die in Nederland actief zijn?
Kan bijvoorbeeld herleid worden waar vandaan de Brabantse nog niet teruggeleide besmettingen komen?
Van de week bij de technische briefing heeft Jaap van Dissel aangegeven dat je op deze manier inderdaad kunt achterhalen waar besmettingen vandaan komen. Dit kun je helaas wel pas achteraf doen. Een van de dingen die hij opnoemde is dat je op deze manier kunt zien of besmettingen van 1 bron komen (bijvoorbeeld veel besmettingen tijdens carnaval), of dat er juist steeds besmettingen komen uit andere gebieden (mensen die in Italië zijn geweest).quote:
Ik vind dit een super interessant topic, hulde voor het aanmaken ervan!
Nu een paar vragen, want ik weet weinig van biologie.
Wat kunnen we met deze informatie? Is er bijvoorbeeld uit te destilleren wat de eigenschappen zijn van de takken die in Nederland actief zijn?
Kan bijvoorbeeld herleid worden waar vandaan de Brabantse nog niet teruggeleide besmettingen komen?

Mijn lekenmening.. De eigenschappen niet, wat je wel zou kunnen zien is als er op een gegeven moment duidelijk blijkt dat regio's andere symptomen vertonen, of er een andere variant van het virus is ontstaan.quote:
Ik vind dit een super interessant topic, hulde voor het aanmaken ervan!
Nu een paar vragen, want ik weet weinig van biologie.
Wat kunnen we met deze informatie? Is er bijvoorbeeld uit te destilleren wat de eigenschappen zijn van de takken die in Nederland actief zijn?
Kan bijvoorbeeld herleid worden waar vandaan de Brabantse nog niet teruggeleide besmettingen komen?
Italie heeft bijna geen data aangeleverd, dus is het niet terug te zien via het genoom.
Hoop dat @Bosbeetle je verder kan helpen.

Ik denk (vrijwel ongeïnformeerd behalve op dat stukje tekst dat op de site staat) dat je nog niet echt van aparte strains kunt spreken maar zo wel kunt zien welke mutaties langzaam accumuleren.
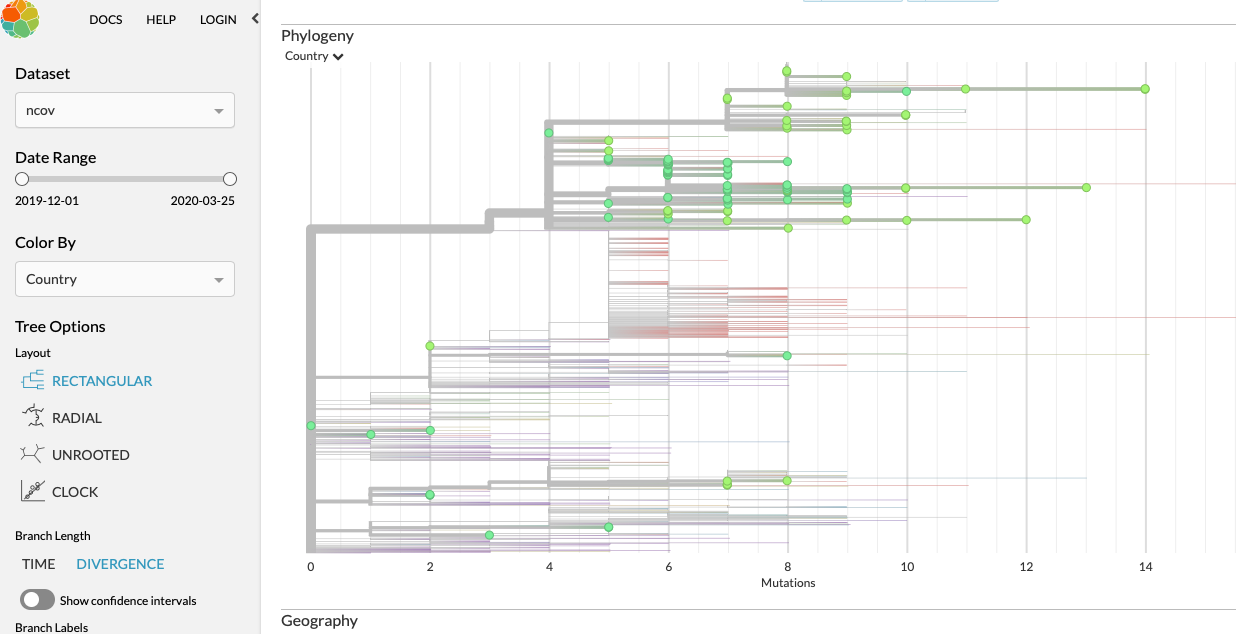
De tabel geeft de meeste info als je hem op divergence zet. Dan zie je op de x-as hoe veel ze uit elkaar liggen en op de y-as hoe ze relateren.
https://nextstrain.org/ncov?c=country&d=tree&m=div&r=country
Deze instelling vind ik het meest informatief.
Wat je nu ziet is dat er in nederland één italiaanse tak is :
https://nextstrain.org/nc(...):A2a&m=div&r=country
En één die losstaat en meer op het wuhan virus lijkt
https://nextstrain.org/ncov?c=country&d=tree&m=div&r=country
De tabel geeft de meeste info als je hem op divergence zet. Dan zie je op de x-as hoe veel ze uit elkaar liggen en op de y-as hoe ze relateren.
https://nextstrain.org/ncov?c=country&d=tree&m=div&r=country
Deze instelling vind ik het meest informatief.
Wat je nu ziet is dat er in nederland één italiaanse tak is :
https://nextstrain.org/nc(...):A2a&m=div&r=country
En één die losstaat en meer op het wuhan virus lijkt
https://nextstrain.org/ncov?c=country&d=tree&m=div&r=country
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Dit is ongeveer hoe zo'n boom gemaakt wordt:
Als je je twee virussen voorstelt met genetische code
AGCUAGCU en er veranderd 1 nucleotide bijvoorbeeld de eerste G dan krijg je een vertakking
als er dan weer twee veranderen maar afzonderlijk bijvoorbeeld de tweede G dan hebben ze een common ancestor
Veranderd nu de eerste G niet maar de tweede wel dan is dat een nieuwe tak
Als je je twee virussen voorstelt met genetische code
AGCUAGCU en er veranderd 1 nucleotide bijvoorbeeld de eerste G dan krijg je een vertakking
| 1 | AGCUAGCU ---- AACUAGCU |
als er dan weer twee veranderen maar afzonderlijk bijvoorbeeld de tweede G dan hebben ze een common ancestor
| 1 2 3 | ------------ AACUAACU AGCUAGCU ---- AACUAGCU ----- ------------ AACUACCU |
Veranderd nu de eerste G niet maar de tweede wel dan is dat een nieuwe tak
| 1 2 3 4 | |------------ AACUAACU AGCUAGCU ---- AACUAGCU ----- | |------------ AACUACCU | ---- AGCUAACU |
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
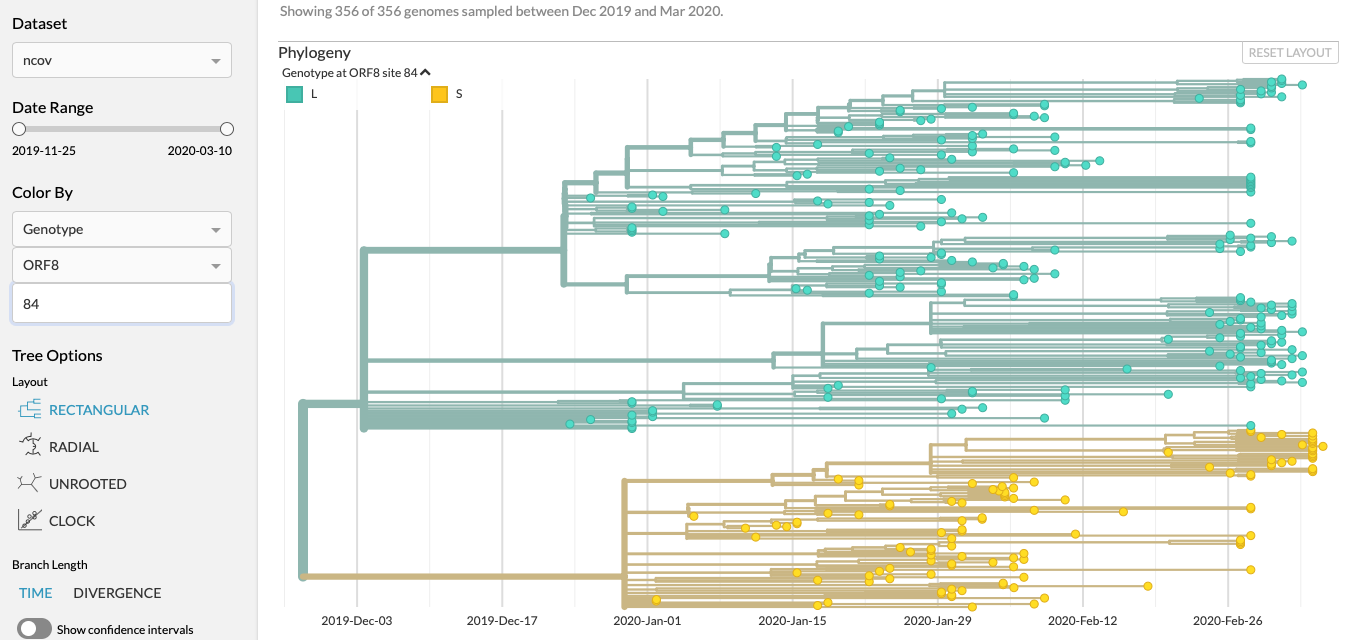
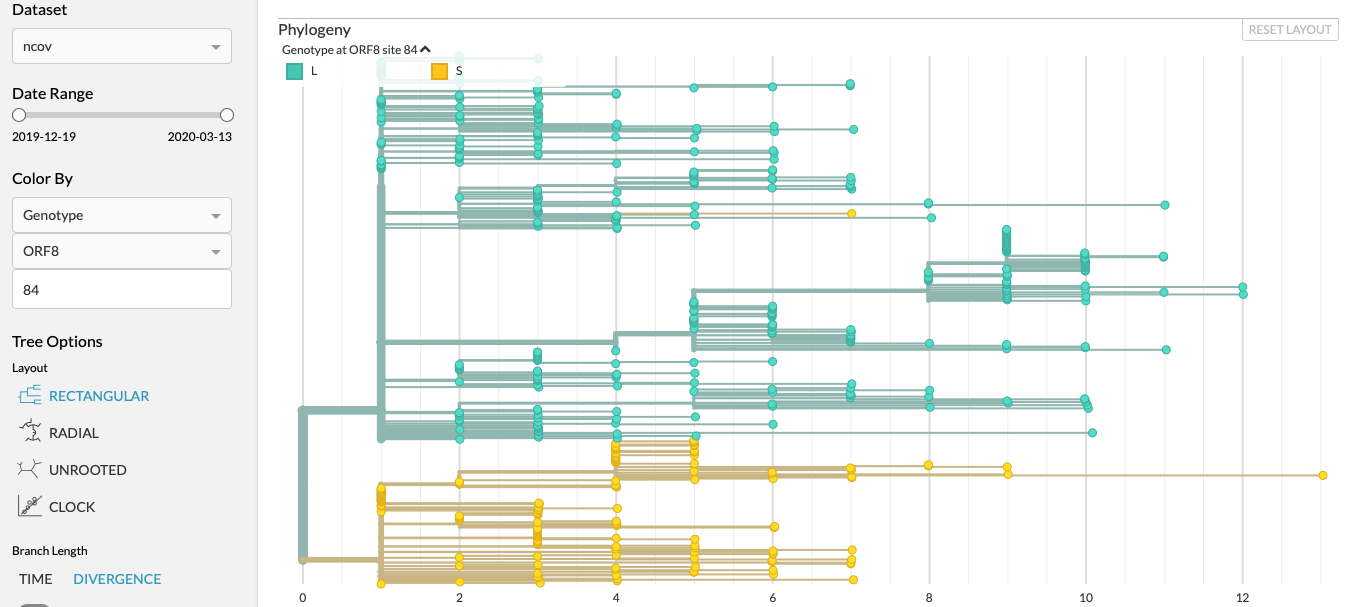
Dat er over een S en L variant gesproken wordt dat betekent dus dat er een Serine (S) of Leucine (L) op positie 84 in het eiwit gecodeerd door ORF8 (open reading frame 8). Dat kan op door verschillende mutaties op RNA niveau komen, want RNA wordt in tripletjes afgelezen (zogenaamde codons).
[ Bericht 10% gewijzigd door Bosbeetle op 12-03-2020 18:37:48 ]
[ Bericht 10% gewijzigd door Bosbeetle op 12-03-2020 18:37:48 ]
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
https://onlinelibrary.wiley.com/doi/pdf/10.1002/jmv.25700
In het kort: het ding muteert niet zoveelquote:In conclusion, our analysis confirms low variability within the
new epidemic virus 2019‐nCoV sequenced specimens, while
highlighting at least two nucleotide positions of higher variability
within protein‐coding regions, and specific amino acid divergences
compared to BCoVs and SARS
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Hey de L naar S in Orf8 pos84 mutatie heeft nog een keer paatsgevonden
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
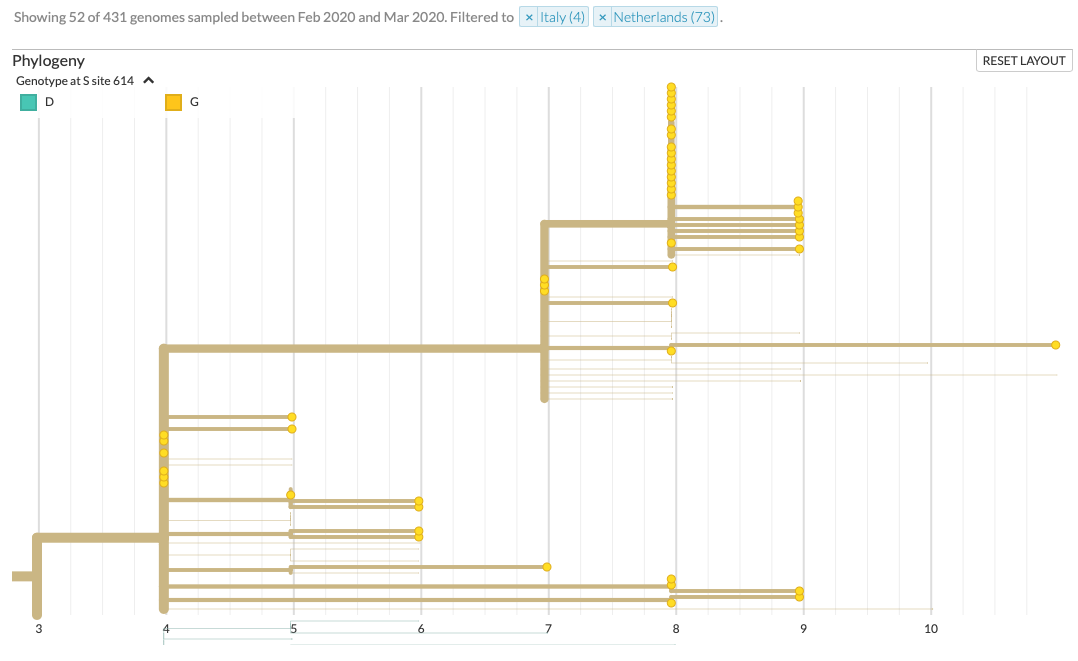
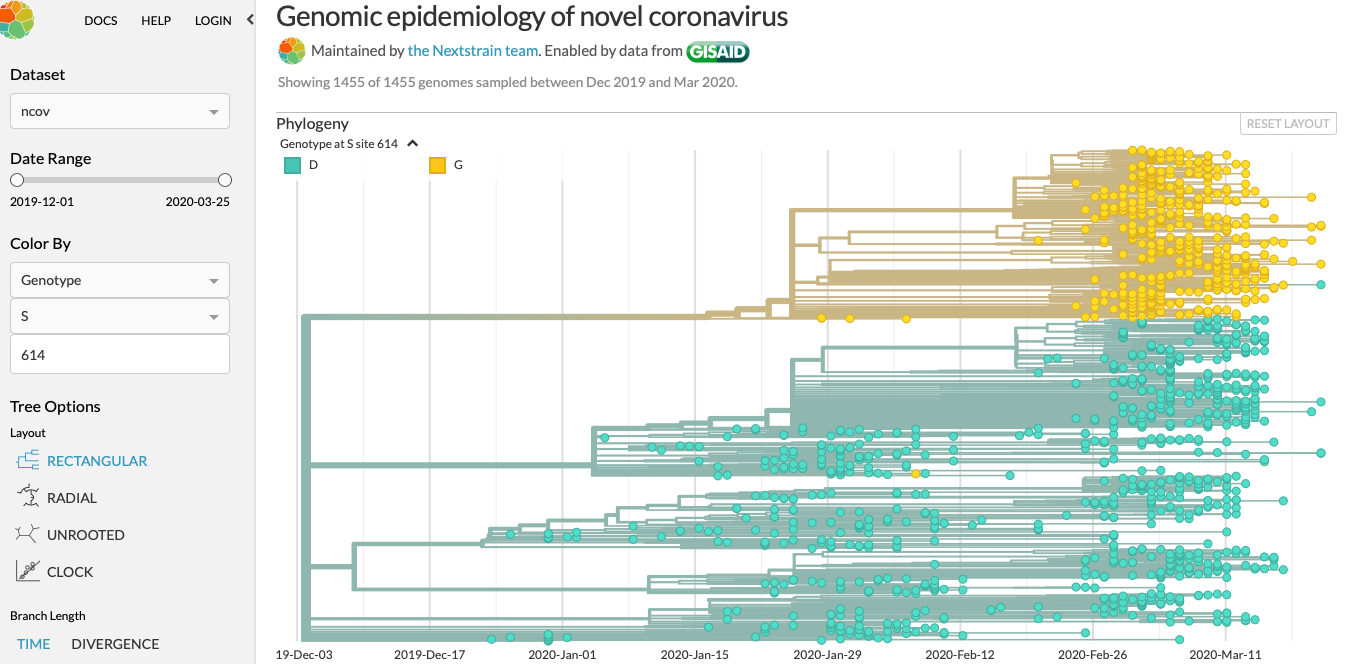
https://nextstrain.org/ncov?c=gt-S_614&m=div
De D naar G in de spike protein op positie 614 lijkt de italiaanse/nederlandse variant te zijn.
De D naar G in de spike protein op positie 614 lijkt de italiaanse/nederlandse variant te zijn.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna

Ik zag het. Er lijken meerdere splitsingen te ontstaan ook. Denk dat er flink wat data is aangevuld.quote:Op vrijdag 13 maart 2020 09:47 schreef Bosbeetle het volgende:
Hey de L naar S in Orf8 pos84 mutatie heeft nog een keer paatsgevonden
quote:
https://nextstrain.org/ncov?c=gt-S_614&m=div
De D naar G in de spike protein op positie 614 lijkt de italiaanse/nederlandse variant te zijn.
Dit geeft wel ineens een heel ander beeld van hoe "onze stamboom" is opgebouwd.
Dit is data tot 8 maart. En ik denk dat ze alleen variaties opsturen en niet gewoon alles. Hier kun je dus alleen vinden als virussen verschillen van anderen. Je zult hier dus niet nederlanders zien staan met een bekende wuhan variant.quote:Op vrijdag 13 maart 2020 16:15 schreef _I het volgende:
[..]
[ afbeelding ]
Dit geeft wel ineens een heel ander beeld van hoe "onze stamboom" is opgebouwd.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna

Ah. Hm. Ow.quote:
[..]
Dit is data tot 8 maart. En ik denk dat ze alleen variaties opsturen en niet gewoon alles. Hier kun je dus alleen vinden als virussen verschillen van anderen. Je zult hier dus niet nederlanders zien staan met een bekende wuhan variant.
Kan ik dan aannemen dat als wij wel 1 op 1, zeg overdreven, alle amerikanen besmet hebben, en al deze virussen komen overeen, dan zouden we die NL'rs niet als groene puntjes terugvinden in het overzicht.
Voor mijn hoofd; datapunten zijn unieke mutaties, geen geïnfecteerden?
[ Bericht 0% gewijzigd door _I op 13-03-2020 16:36:51 ]
Aantal geïnfecteerden staan wel op de kaart maar niet in de puntjes, geloof ik.quote:Op vrijdag 13 maart 2020 16:27 schreef _I het volgende:
[..]
Ah. Hm. Ow.
Kan ik dan aannemen dat als wij wel 1 op 1, zeg overdreven, alle amerikanen besmet hebben, en al deze virussen komen overeen, dan zouden we die NL'rs niet als groene puntjes terugvinden in het overzicht.
Voor mijn hoofd; datapunten zijn unieke mutaties, geen geïnfecteerden?
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Bon!quote:
[..]
Aantal geïnfecteerden staan wel op de kaart maar niet in de puntjes, geloof ik.
Je ziet in nederland dan ook taart diagrammen bij die S pos614 mutatie.quote:
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Dus stel als het virus iemand met een chromosoomafwijking besmet, zou het virus zichzelf dan verder gaan ontwikkelen?
Google is your friend, abuse your friends
Had ik maar biologie gestudeerd.quote:
[..]
Je ziet in nederland dan ook taart diagrammen bij die S pos614 mutatie.
Dat veranderd niets aan de besmetting, een virus heeft wat tools aan boord en kaapt de machinerie van zijn gastheer.quote:
Dus stel als het virus iemand met een chromosoomafwijking besmet, zou het virus zichzelf dan verder gaan ontwikkelen?
In het kort, het virus brengt RNA in gastheer cellen, die gastheer ziet het als normaal RNA leest het af en maakt de bouwstenen (eiwit) voor nieuw virus.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
De basis hiervan zit toch wel in biologie op de middelbare school.quote:
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Daar heb ik het heel snel laten varen. Scheikunde, biologie en natuurkunde lieten me de tijd vergeten.quote:
[..]
De basis hiervan zit toch wel in biologie op de middelbare school.
Als ik het allemaal over mocht doen...
Cross post
COR / Nextstrain
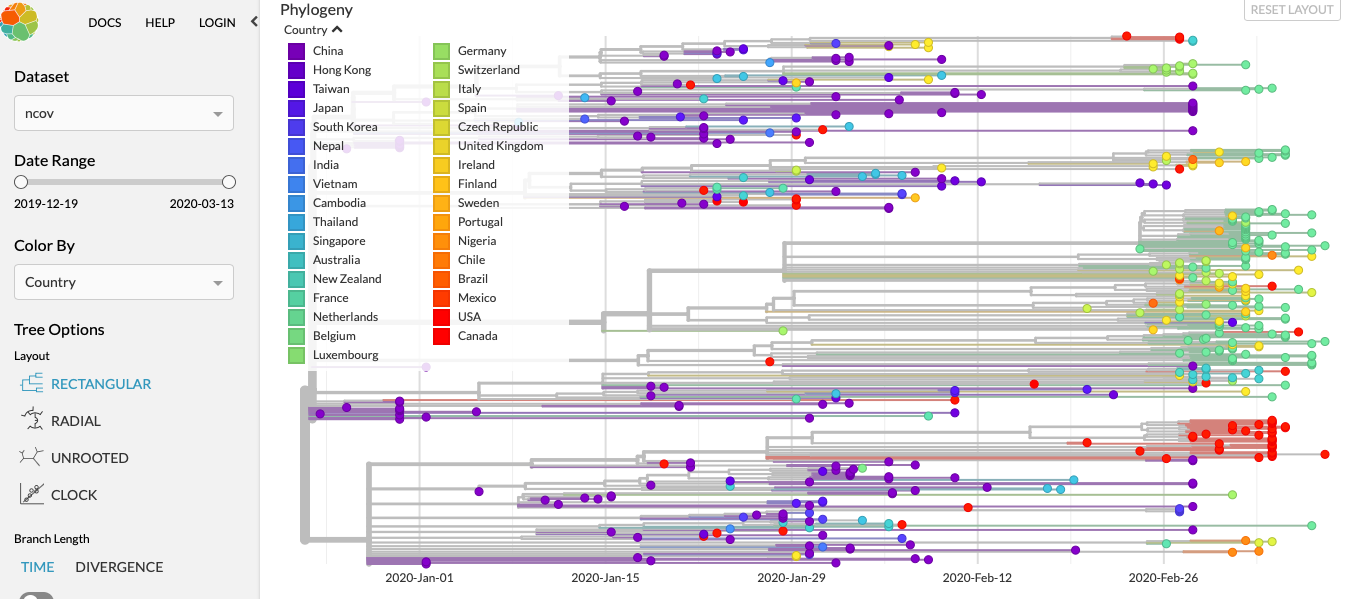
Nextstrain is een open-source project, waarbij samples worden verzameld van mensen die het virus hebben en door de RNA-sequenties met elkaar te vergelijken is het mogelijk om te zien welk sample voortkomt uit welk ander sample. Net als het plotten van de oorsprong van de mens en de relatie tot de apen.
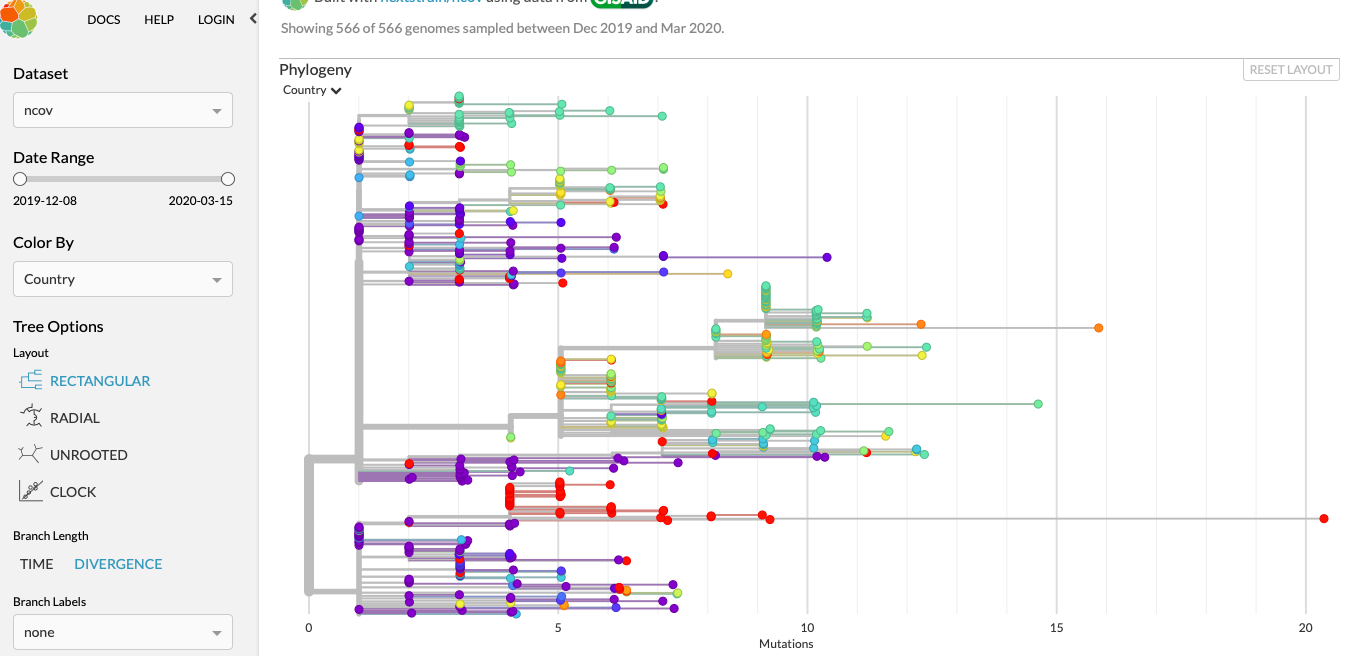
Voor elk land is een kleur en de grafiek is uitgezet in tijd. Wij zitten in de "groene hoek" en zo kan je zien hoe "ons" virus gelinkt is aan het virus wat in de US overheerst (rood)
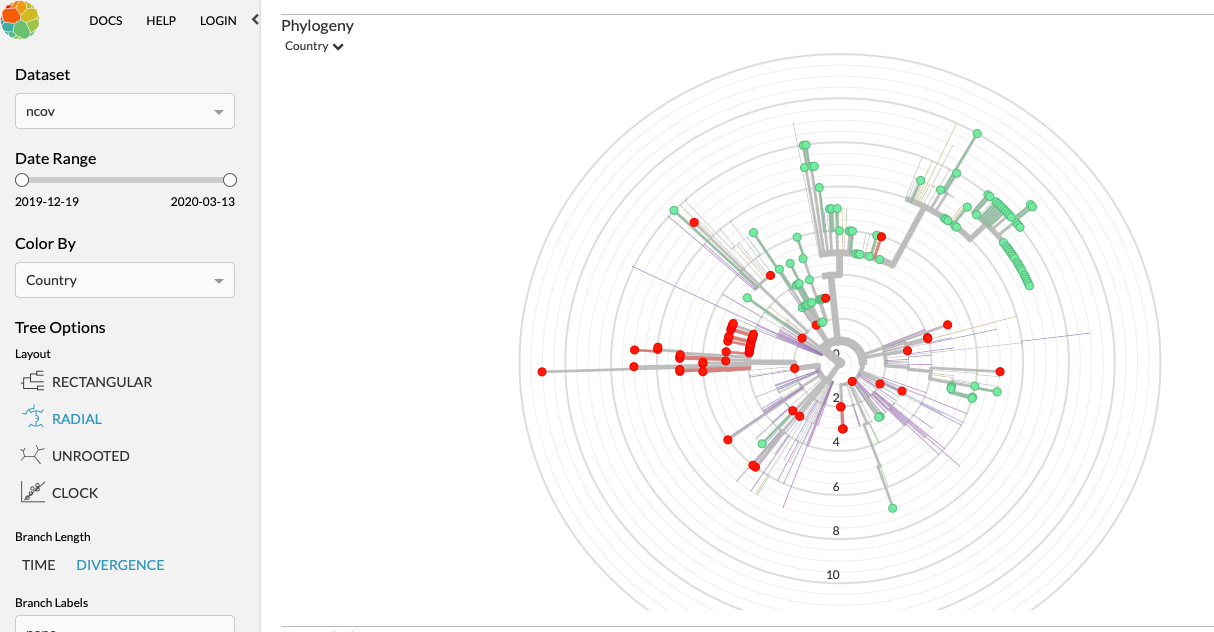
Als je de data niet presenteert in 'tijd', maar in 'divergence' dat wordt in kaart gebracht hoeveel "afwijkingen" de samples hebben t.o.v. elkaar.
Voor de US (rood) zie je dan dat er een zieke is, die meer dan 12 mutaties heeft van de eerste gevonden RNA.
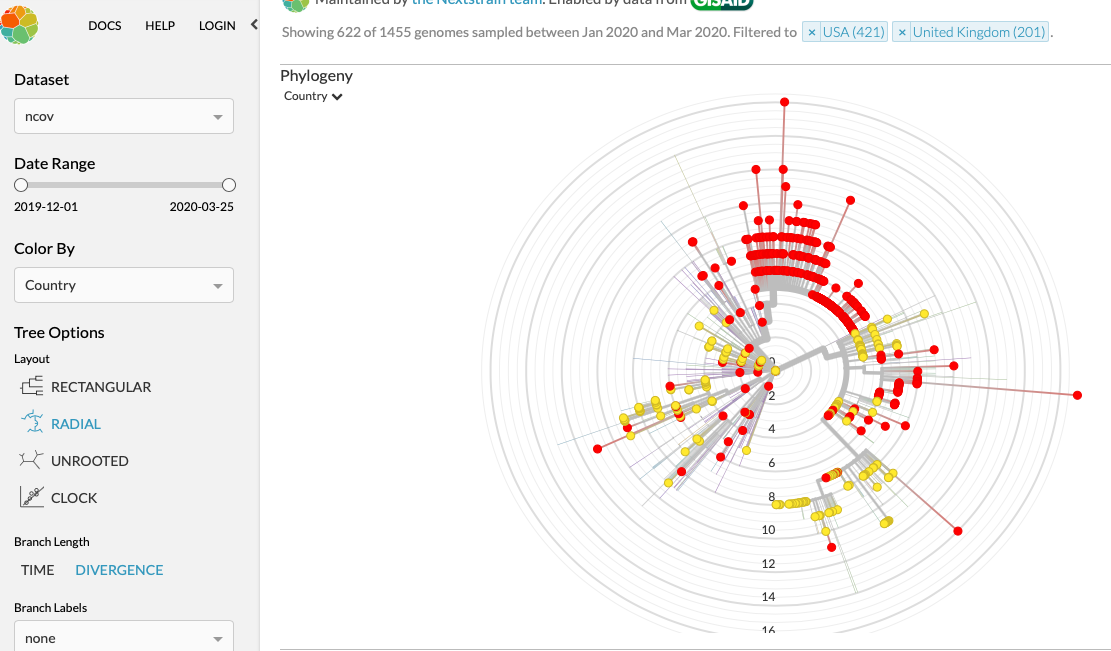
In een taartdiagram, gefilterd voor alleen NL en US, zie je redelijk helder dat er weinig vermenging is. Er zit een enkel rood puntje tussen de groene in en vice versa.
Als je filtert op de US en China, dan zie je dat bijna alle rode puntjes voortkomen uit paarse. De besmettingen in de US komen dus logischerwijs voort uit China en niet uit onze regio. Je ziet dat die nu heel licht grijs zijn en los staat van het gekleurde gedeelte
Wetenschappers zagen dat op een bepaald genoom er een mutaties was ontstaan, de L en de S; hier geel en blauw.
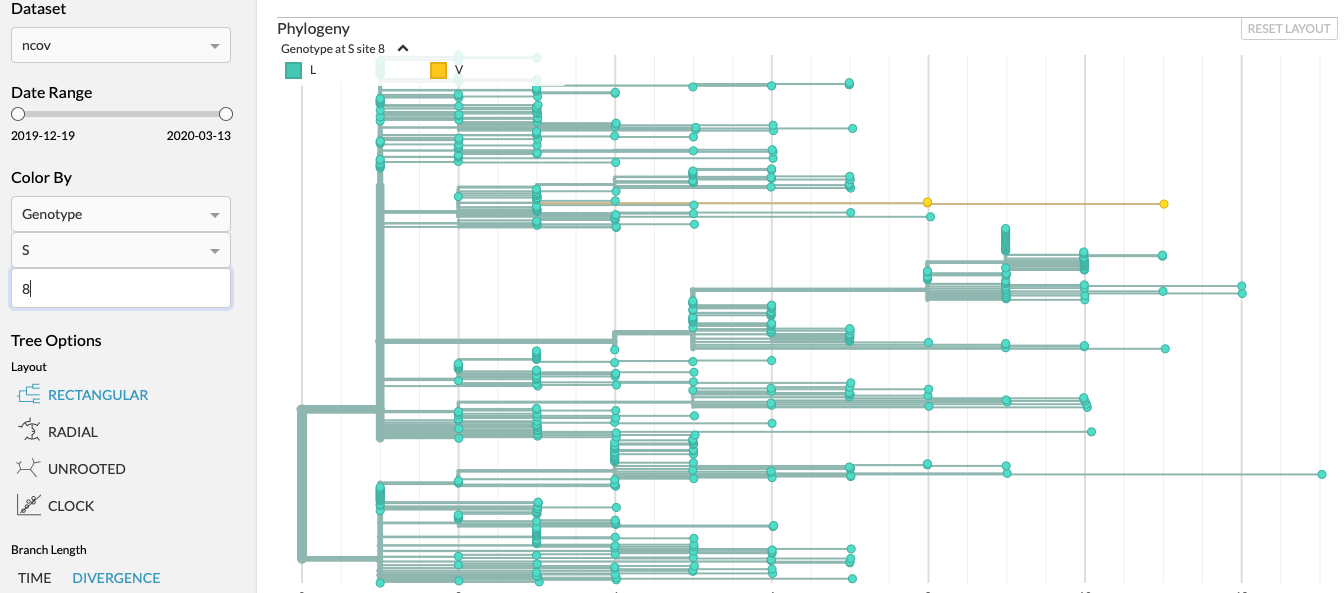
Ik vond gisteren zelf nog zo'n mutatie in een ander genoom (bij toeval)
En Bosbeetle wees me op nog een andere;
Deze mutaties maken het mogelijk om het verloop in kaart te brengen van het virus in tijd, maar voor wat men nu weet zitten deze mutaties niet in die genomen die zorgen voor mutaties in het virus zelf. Er is dus nog niet te zeggen of de ene mutatie agressiever is dan de ander, daarvoor was de sample-size te klein en de verwachtinig is dus dat het niet-coderende genomen zijn, die dus het virus niet veranderen in effect.
Zit er ook niet diep in, maar dit is wat ik ervan begrepen heb.
Er loopt ook een topic over, maar zal het proberen uit te leggen;quote:
Ok thanks. Ergens in dit topic is hier namelijk een uitgebreide analyse van gedaan. Inclusief waar deze strains heen zijn gegaan. Die analyse toonde aan dat de L en S strain in China zijn ontstaan, maar vooral de L-strain naar Europa en Amerika zijn gegaan. Niet waar dus?
COR / Nextstrain
Nextstrain is een open-source project, waarbij samples worden verzameld van mensen die het virus hebben en door de RNA-sequenties met elkaar te vergelijken is het mogelijk om te zien welk sample voortkomt uit welk ander sample. Net als het plotten van de oorsprong van de mens en de relatie tot de apen.
Voor elk land is een kleur en de grafiek is uitgezet in tijd. Wij zitten in de "groene hoek" en zo kan je zien hoe "ons" virus gelinkt is aan het virus wat in de US overheerst (rood)
Als je de data niet presenteert in 'tijd', maar in 'divergence' dat wordt in kaart gebracht hoeveel "afwijkingen" de samples hebben t.o.v. elkaar.
Voor de US (rood) zie je dan dat er een zieke is, die meer dan 12 mutaties heeft van de eerste gevonden RNA.

In een taartdiagram, gefilterd voor alleen NL en US, zie je redelijk helder dat er weinig vermenging is. Er zit een enkel rood puntje tussen de groene in en vice versa.
Als je filtert op de US en China, dan zie je dat bijna alle rode puntjes voortkomen uit paarse. De besmettingen in de US komen dus logischerwijs voort uit China en niet uit onze regio. Je ziet dat die nu heel licht grijs zijn en los staat van het gekleurde gedeelte
Wetenschappers zagen dat op een bepaald genoom er een mutaties was ontstaan, de L en de S; hier geel en blauw.
Ik vond gisteren zelf nog zo'n mutatie in een ander genoom (bij toeval)
En Bosbeetle wees me op nog een andere;

Deze mutaties maken het mogelijk om het verloop in kaart te brengen van het virus in tijd, maar voor wat men nu weet zitten deze mutaties niet in die genomen die zorgen voor mutaties in het virus zelf. Er is dus nog niet te zeggen of de ene mutatie agressiever is dan de ander, daarvoor was de sample-size te klein en de verwachtinig is dus dat het niet-coderende genomen zijn, die dus het virus niet veranderen in effect.
Zit er ook niet diep in, maar dit is wat ik ervan begrepen heb.
Kudo's voor dit topic. Zeer boeiende materie.
The hardware is just a box you buy only because you want to play Mario games - Yamauchi
Mr. Zurkon doesn't need bolts, his currency is pain
Roy-O-Rama | Backlog | Wish-list
Mr. Zurkon doesn't need bolts, his currency is pain
Roy-O-Rama | Backlog | Wish-list
Kan je ook 2x ziek worden van verschillende strains?
"This is your life and it's ending one minute at a time." - Tyler Durden
"Sand is overrated. It's just tiny, little rocks." - Joel
"Sand is overrated. It's just tiny, little rocks." - Joel
ja, zelfs tegelijk van die twee strainsquote:
Kan je ook 2x ziek worden van verschillende strains?
Dat kun je niet zomaar zeggen.quote:
Kan je ook 2x ziek worden van verschillende strains?
Sommige van die afwijkingen. Zijn ‘niet effectief’. Dwz dat ze niet coderen voor ‘functionaliteit ‘.
Daarnaast weet nemen nog niet hoe het zit met immunisatie. Ik lees de wildste verhalen, waarvan de meeste bullshit.
Whatever...
Als je op die staafdiagrammen grafiek kijkt onderin beeld dan zie je daar welke posities het meest muteren, door op zo'n balk te klikken zie je het gelijk in de boom grafiek. Onder de staaf diagram grafiek kun je ook inzoomen en uitzoomen op het hele genoom. Je kunt die ook switchen tussen aminozuur en nucleotide ( eiwit volgorde vs Rna volgorde).quote:
Cross post
[..]
Er loopt ook een topic over, maar zal het proberen uit te leggen;
COR / Nextstrain
Nextstrain is een open-source project, waarbij samples worden verzameld van mensen die het virus hebben en door de RNA-sequenties met elkaar te vergelijken is het mogelijk om te zien welk sample voortkomt uit welk ander sample. Net als het plotten van de oorsprong van de mens en de relatie tot de apen.
Voor elk land is een kleur en de grafiek is uitgezet in tijd. Wij zitten in de "groene hoek" en zo kan je zien hoe "ons" virus gelinkt is aan het virus wat in de US overheerst (rood)
[ afbeelding ]
Als je de data niet presenteert in 'tijd', maar in 'divergence' dat wordt in kaart gebracht hoeveel "afwijkingen" de samples hebben t.o.v. elkaar.
Voor de US (rood) zie je dan dat er een zieke is, die meer dan 12 mutaties heeft van de eerste gevonden RNA.
[ afbeelding ]
In een taartdiagram, gefilterd voor alleen NL en US, zie je redelijk helder dat er weinig vermenging is. Er zit een enkel rood puntje tussen de groene in en vice versa.
[ afbeelding ]
Als je filtert op de US en China, dan zie je dat bijna alle rode puntjes voortkomen uit paarse. De besmettingen in de US komen dus logischerwijs voort uit China en niet uit onze regio. Je ziet dat die nu heel licht grijs zijn en los staat van het gekleurde gedeelte
[ afbeelding ]
Wetenschappers zagen dat op een bepaald genoom er een mutaties was ontstaan, de L en de S; hier geel en blauw.
[ afbeelding ]
Ik vond gisteren zelf nog zo'n mutatie in een ander genoom (bij toeval)
[ afbeelding ]
En Bosbeetle wees me op nog een andere;
[ afbeelding ]
Deze mutaties maken het mogelijk om het verloop in kaart te brengen van het virus in tijd, maar voor wat men nu weet zitten deze mutaties niet in die genomen die zorgen voor mutaties in het virus zelf. Er is dus nog niet te zeggen of de ene mutatie agressiever is dan de ander, daarvoor was de sample-size te klein en de verwachtinig is dus dat het niet-coderende genomen zijn, die dus het virus niet veranderen in effect.
[ afbeelding ]
Zit er ook niet diep in, maar dit is wat ik ervan begrepen heb.
Waarom bepaalde posities meer muteren dan andere kan met selectiedruk te maken hebben, en dat is weer interessant voor mogelijke vaccins.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
quote:
[..]
Als je op die staafdiagrammen grafiek kijkt onderin beeld dan zie je daar welke posities het meest muteren, door op zo'n balk te klikken zie je het gelijk in de boom grafiek. Onder de staaf diagram grafiek kun je ook inzoomen en uitzoomen op het hele genoom. Je kunt die ook switchen tussen aminozuur en nucleotide ( eiwit volgorde vs Rna volgorde).
Waarom bepaalde posities meer muteren dan andere kan met selectiedruk te maken hebben, en dat is weer interessant voor mogelijke vaccins.
Dat zoekt wel een stuk sneller. M 175 ook best apart. Fascinerend.
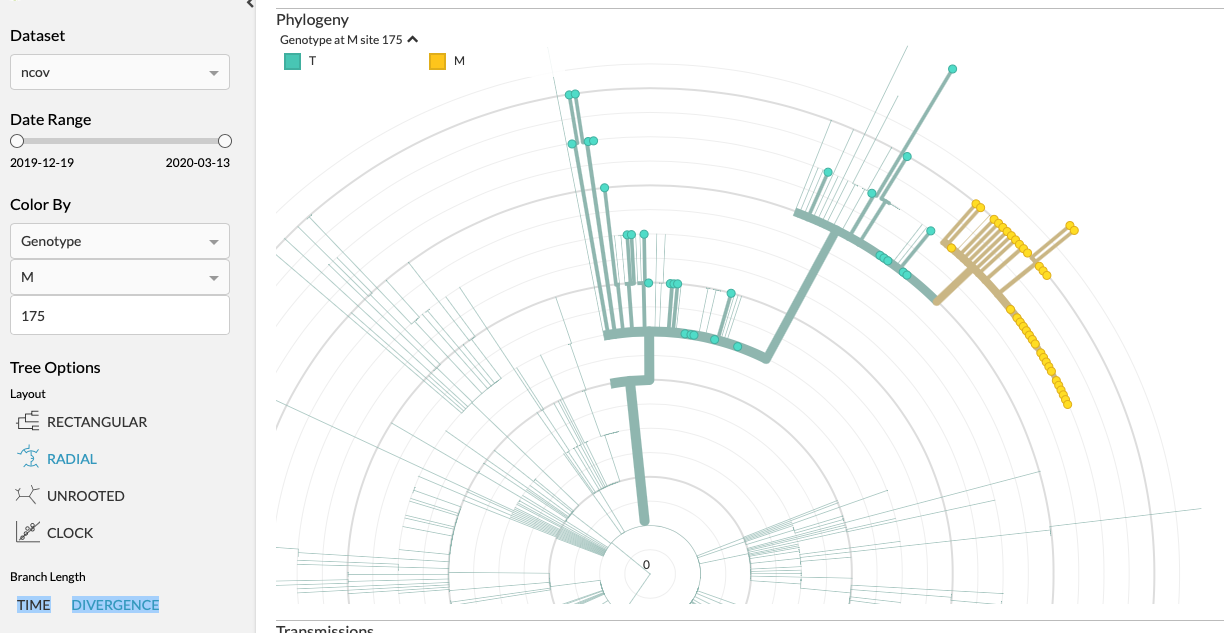
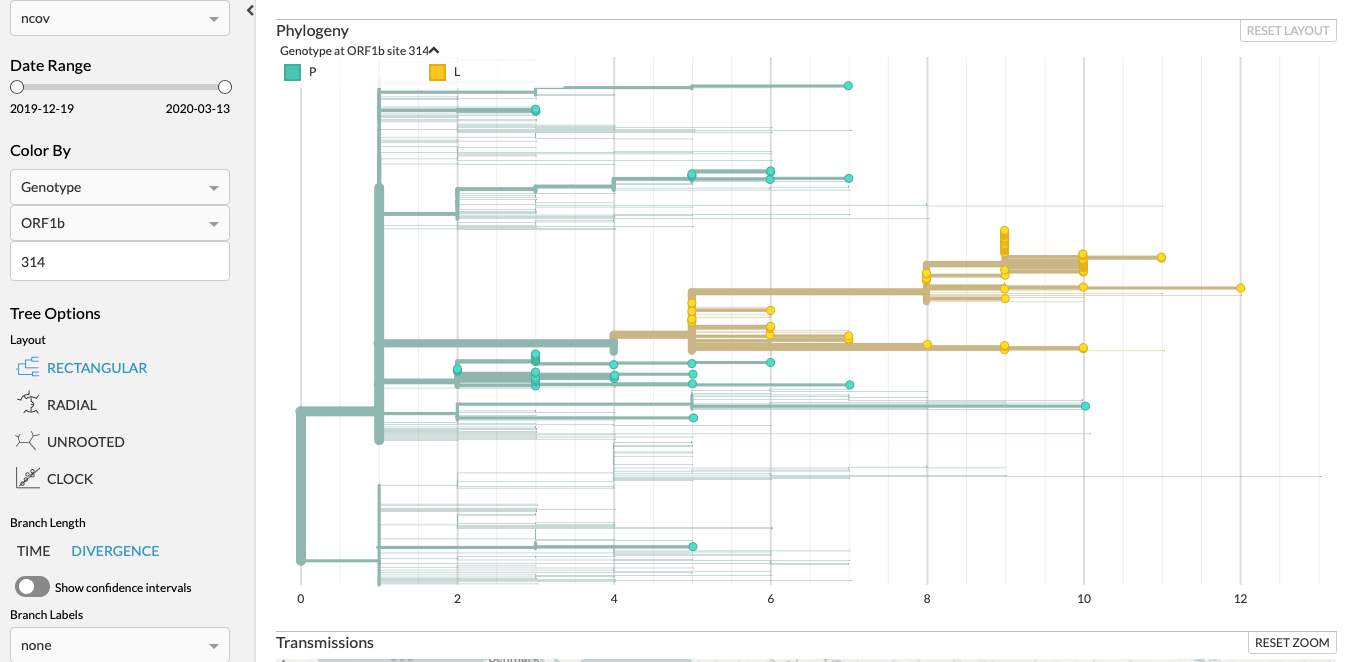
ORF3a 251 heeft ook wel een specifieke tak inmiddels.
Jammer dat het lab geen live verbinding heeft.
Jammer dat het lab geen live verbinding heeft.
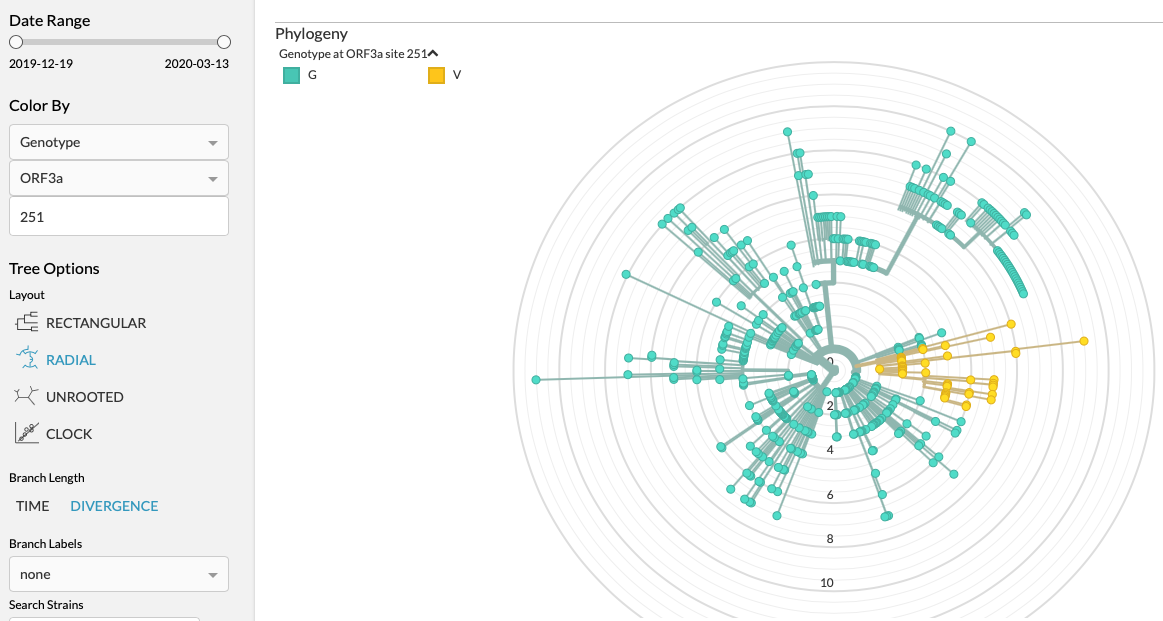
Er zijn er best een aantal en NL zit vaak in de afwijkende groep (filter staat alleen op NL)
Zou wel een stuk helpen als Italie niet maar 4 bolletjes had. Is toch redelijk de "missing link" voor ons.
Zou wel een stuk helpen als Italie niet maar 4 bolletjes had. Is toch redelijk de "missing link" voor ons.
Voor extra info
spike (S) protein, nucleocapsid (N) protein, membrane (M) protein, and the envelope (E) protein dit zijn de strucuterele eiwitten van dit virus en dus nodig om een virus deeltje te maken.
Spike is een receptor waarmee het virus aan gastheer cellen bind. Nucleocapsid is het enige eiwit dan aan het RNA bind en zorgt voor het inpakken van het virus. M is de organisator die het meest contact heeft met de anderen, en interacteert met S,N en E. E is het kleinste eiwit wat wel in het membraan zit maar vooral zijn werkt doet in de gastheer cel.
https://virologyj.biomedc(...)86/s12985-019-1182-0
De eiwitten die gemaakt worden door ORF1-8 zijn niet structureel.
spike (S) protein, nucleocapsid (N) protein, membrane (M) protein, and the envelope (E) protein dit zijn de strucuterele eiwitten van dit virus en dus nodig om een virus deeltje te maken.
Spike is een receptor waarmee het virus aan gastheer cellen bind. Nucleocapsid is het enige eiwit dan aan het RNA bind en zorgt voor het inpakken van het virus. M is de organisator die het meest contact heeft met de anderen, en interacteert met S,N en E. E is het kleinste eiwit wat wel in het membraan zit maar vooral zijn werkt doet in de gastheer cel.
https://virologyj.biomedc(...)86/s12985-019-1182-0
De eiwitten die gemaakt worden door ORF1-8 zijn niet structureel.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Hm, probeer je te volgen.quote:
Voor extra info
spike (S) protein, nucleocapsid (N) protein, membrane (M) protein, and the envelope (E) protein dit zijn de strucuterele eiwitten van dit virus en dus nodig om een virus deeltje te maken.
Spike is een receptor waarmee het virus aan gastheer cellen bind. Nucleocapsid is het enige eiwit dan aan het RNA bind en zorgt voor het inpakken van het virus. M is de organisator die het meest contact heeft met de anderen, en interacteert met S,N en E. E is het kleinste eiwit wat wel in het membraan zit maar vooral zijn werkt doet in de gastheer cel.
https://virologyj.biomedc(...)86/s12985-019-1182-0
De eiwitten die gemaakt worden door ORF1-8 zijn niet structureel.
Welke mutaties zijn dan wel structureel en achter welke mutaties schuilt "gevaar"? Is dat te duiden?
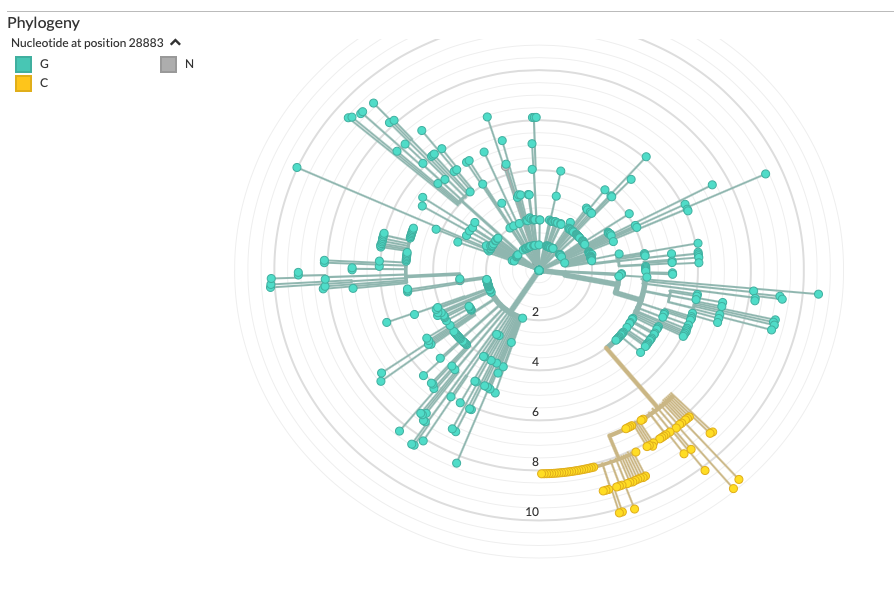
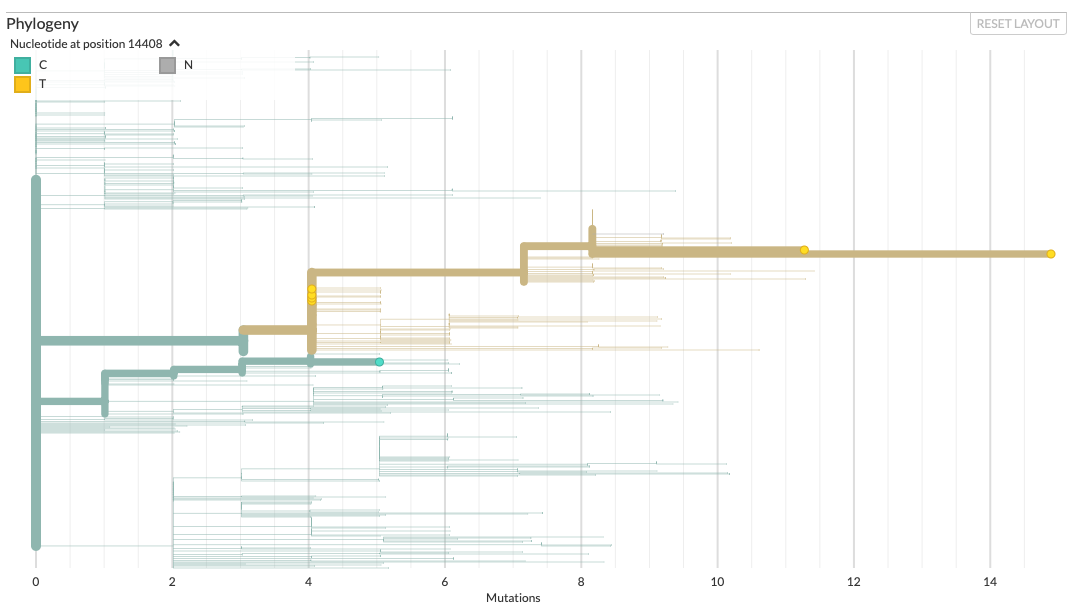
https://nextstrain.org/ncov?c=gt-nuc_28883&m=div
Zoals die mutatie op N, zou dat iets structureels kunnen zijn?

Heb de link doorgenomen, maar moet ergens mn verlies erkennen. Denk dat dit het punt is.
De genoemde eiwitten zorgen voor opbouw en 'sammenstellen' van het virus. Het zegt niets over gevaar.quote:Op zondag 15 maart 2020 11:05 schreef _I het volgende:
[..]
Hm, probeer je te volgen.
Welke mutaties zijn dan wel structureel en achter welke mutaties schuilt "gevaar"? Is dat te duiden?
https://nextstrain.org/ncov?c=gt-nuc_28883&m=div
Zoals die mutatie op N, zou dat iets structureels kunnen zijn?
[ afbeelding ]
Heb de link doorgenomen, maar moet ergens mn verlies erkennen. Denk dat dit het punt is.
Je kijkt hier eigenlijk naar evolutie aan het werk, veranderd er iets in bijvoorbeeld het spike eiwit waardoor het virus iets snellen binnenkomt, dan zal dat virus iets voorsprong krijgen en steeds vaker voorkomen. Maar dat kan ook gebeuren als het virus iets 'milder' wordt en men het makkelijker ongemerkt doorgeeft. Of als een mutatie zorgt voor veel meer hoesten.
Uit het artikel gaat het vooral om dit stuk dat goed uitlegt hoe het virus in elkaar zit:
quote:The coronaviral genome encodes four major structural proteins: the spike (S) protein, nucleocapsid (N) protein, membrane (M) protein, and the envelope (E) protein, all of which are required to produce a structurally complete viral particle [29, 37, 38]. More recently, however, it has become clear that some CoVs do not require the full ensemble of structural proteins to form a complete, infectious virion, suggesting that some structural proteins might be dispensable or that these CoVs might encode additional proteins with overlapping compensatory functions [35, 37, 39,40,41,42]. Individually, each protein primarily plays a role in the structure of the virus particle, but they are also involved in other aspects of the replication cycle. The S protein mediates attachment of the virus to the host cell surface receptors and subsequent fusion between the viral and host cell membranes to facilitate viral entry into the host cell [42,43,44]. In some CoVs, the expression of S at the cell membrane can also mediate cell-cell fusion between infected and adjacent, uninfected cells. This formation of giant, multinucleated cells, or syncytia, has been proposed as a strategy to allow direct spreading of the virus between cells, subverting virus-neutralising antibodies [45,46,47].
Unlike the other major structural proteins, N is the only protein that functions primarily to bind to the CoV RNA genome, making up the nucleocapsid [48]. Although N is largely involved in processes relating to the viral genome, it is also involved in other aspects of the CoV replication cycle and the host cellular response to viral infection [49]. Interestingly, localisation of N to the endoplasmic reticulum (ER)-Golgi region has proposed a function for it in assembly and budding [50, 51]. However, transient expression of N was shown to substantially increase the production of virus-like particles (VLPs) in some CoVs, suggesting that it might not be required for envelope formation, but for complete virion formation instead [41, 42, 52, 53].
The M protein is the most abundant structural protein and defines the shape of the viral envelope [54]. It is also regarded as the central organiser of CoV assembly, interacting with all other major coronaviral structural proteins [29]. Homotypic interactions between the M proteins are the major driving force behind virion envelope formation but, alone, is not sufficient for virion formation [54,55,56]. Interaction of S with M is necessary for retention of S in the ER-Golgi intermediate compartment (ERGIC)/Golgi complex and its incorporation into new virions, but dispensable for the assembly process [37, 45, 57]. Binding of M to N stabilises the nucleocapsid (N protein-RNA complex), as well as the internal core of virions, and, ultimately, promotes completion of viral assembly [45, 58, 59]. Together, M and E make up the viral envelope and their interaction is sufficient for the production and release of VLPs [37, 60,61,62,63,64].
The E protein is the smallest of the major structural proteins, but also the most enigmatic. During the replication cycle, E is abundantly expressed inside the infected cell, but only a small portion is incorporated into the virion envelope [65]. The majority of the protein is localised at the site of intracellular trafficking, viz. the ER, Golgi, and ERGIC, where it participates in CoV assembly and budding [66]. Recombinant CoVs have lacking E exhibit significantly reduced viral titres, crippled viral maturation, or yield propagation incompetent progeny, demonstrating the importance of E in virus production and maturation [35, 39, 40, 67, 68].
[ Bericht 5% gewijzigd door Bosbeetle op 15-03-2020 11:58:19 ]
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Dank! Het roept alleen zoveel vragen op, dus lijkt het me beter om het los te laten.quote:
[..]
De genoemde eiwitten zorgen voor opbouw en 'sammenstellen' van het virus. Het zegt niets over gevaar.
Je kijkt hier eigenlijk naar evolutie aan het werk, veranderd er iets in bijvoorbeeld het spike eiwit waardoor het virus iets snellen binnenkomt, dan zal dat virus iets voorsprong krijgen en steeds vaker voorkomen. Maar dat kan ook gebeuren als het virus iets 'milder' wordt en men het makkelijker ongemerkt doorgeeft. Of als een mutatie zorgt voor veel meer hoesten.
Uit het artikel gaat het vooral om dit stuk dat goed uitlegt hoe het virus in elkaar zit:
[..]
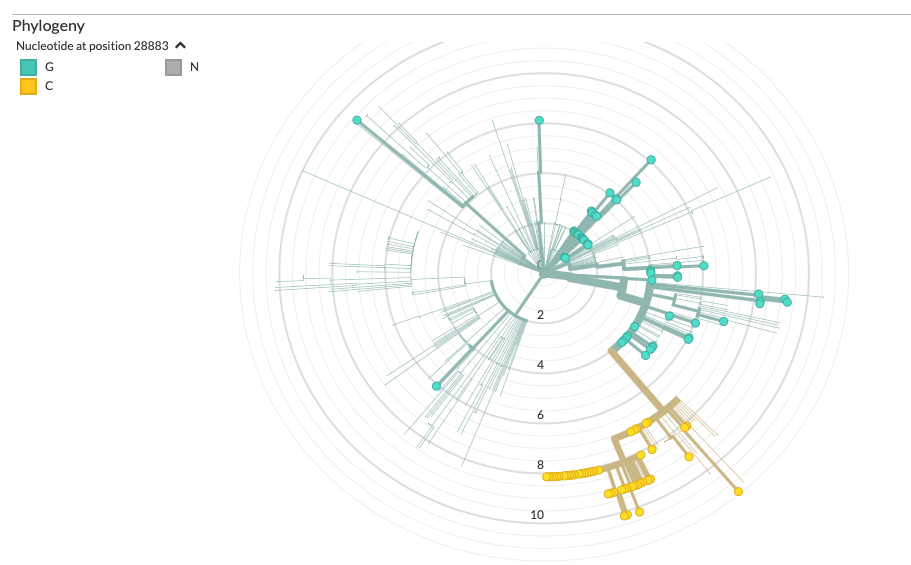
Het valt me namelijk op dat NL er vaak uitspringt;
Alles
Alleen NL
Afwijkingen in patronen zijn als een soort heroine voor me. Heb wel even genoeg gehad de afgelopen weken.
Maar waar dienen ze dan voor?quote:
De eiwitten die gemaakt worden door ORF1-8 zijn niet structureel.
A Robin Redbreast in a Cage Puts all Heaven in a Rage.
Het feit dat er zo'n tak is betekent al dat daar specifieke mutaties zitten die tak is verwant maar niet aan de rest, je kunt die voor alle takken vinden. Omdat die takken structuur daar op gebaseerd is!quote:Op zondag 15 maart 2020 19:21 schreef _I het volgende:
[..]
Dank! Het roept alleen zoveel vragen op, dus lijkt het me beter om het los te laten.
Het valt me namelijk op dat NL er vaak uitspringt;
Alles
[ afbeelding ]
Alleen NL
[ afbeelding ]
Afwijkingen in patronen zijn als een soort heroine voor me. Heb wel even genoeg gehad de afgelopen weken.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Geen idee maar bijvoorbeeld proteasen (eiwitten die andere eiwitten kunnen afbreken) of andere eiwitten met onbekende functies. Ik ben er niet zover in gedoken, die protease heb ik horen noemen.quote:
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna

Abstract
SARS-CoV-2 is a betacoronavirus that is responsible for the COVID-19 pandemic. The genome of SARS-CoV-2 was reported recently, but its transcriptomic architecture is unknown. Utilizing two complementary sequencing techniques, we here present a high-resolution map of the SARS-CoV-2 transcriptome and epitranscriptome.
DNA nanoball sequencing shows that the transcriptome is highly complex owing to numerous recombination events, both canonical and noncanonical. In addition to the genomic RNA and subgenomic RNAs common in all coronaviruses, SARS-CoV-2 produces a large number of transcripts encoding unknown ORFs with fusion, deletion, and/or frameshift.
Using nanopore direct RNA sequencing, we further find at least 41 RNA modification sites on viral transcripts, with the most frequent motif being AAGAA. Modified RNAs have shorter poly(A) tails than unmodified RNAs, suggesting a link between the internal modification and the 3′ tail. Functional investigation of the unknown ORFs and RNA modifications discovered in this study will open new directions to our understanding of the life cycle and pathogenicity of SARS-CoV-2.
SARS-CoV-2 is a betacoronavirus that is responsible for the COVID-19 pandemic. The genome of SARS-CoV-2 was reported recently, but its transcriptomic architecture is unknown. Utilizing two complementary sequencing techniques, we here present a high-resolution map of the SARS-CoV-2 transcriptome and epitranscriptome.
DNA nanoball sequencing shows that the transcriptome is highly complex owing to numerous recombination events, both canonical and noncanonical. In addition to the genomic RNA and subgenomic RNAs common in all coronaviruses, SARS-CoV-2 produces a large number of transcripts encoding unknown ORFs with fusion, deletion, and/or frameshift.
Using nanopore direct RNA sequencing, we further find at least 41 RNA modification sites on viral transcripts, with the most frequent motif being AAGAA. Modified RNAs have shorter poly(A) tails than unmodified RNAs, suggesting a link between the internal modification and the 3′ tail. Functional investigation of the unknown ORFs and RNA modifications discovered in this study will open new directions to our understanding of the life cycle and pathogenicity of SARS-CoV-2.
Kleine cartoon waarom het RNA van het virus niet één op één de code is voor de eiwitten die het produceert:quote:Op maandag 16 maart 2020 07:27 schreef _I het volgende:
Abstract
SARS-CoV-2 is a betacoronavirus that is responsible for the COVID-19 pandemic. The genome of SARS-CoV-2 was reported recently, but its transcriptomic architecture is unknown. Utilizing two complementary sequencing techniques, we here present a high-resolution map of the SARS-CoV-2 transcriptome and epitranscriptome.
DNA nanoball sequencing shows that the transcriptome is highly complex owing to numerous recombination events, both canonical and noncanonical. In addition to the genomic RNA and subgenomic RNAs common in all coronaviruses, SARS-CoV-2 produces a large number of transcripts encoding unknown ORFs with fusion, deletion, and/or frameshift.
Using nanopore direct RNA sequencing, we further find at least 41 RNA modification sites on viral transcripts, with the most frequent motif being AAGAA. Modified RNAs have shorter poly(A) tails than unmodified RNAs, suggesting a link between the internal modification and the 3′ tail. Functional investigation of the unknown ORFs and RNA modifications discovered in this study will open new directions to our understanding of the life cycle and pathogenicity of SARS-CoV-2.
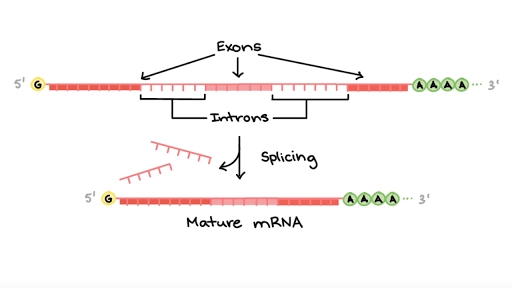
Dit is een simpel voorbeeld van een gen bij een virus zit het wat lastiger. Maar je ziet hier de genetische code (die bij dit virus al bestaat uit RNA) maar daarna wordt dat RNA geprocessed om zo tot stukken te komen die vervolgens worden afgelezen en vertaald naar eiwit.
Het stuk dat I_ deelt laat zien dat je met 1 stuk RNA toch veel verschillende RNA transcripten kunt krijgen, bijvoorbeeld door deleties (kleine stukken die verdwijnen) of recombnatie (stukken die aan elkaar geplakt worden die in het basis RNA niet bij elkaar zitten) of frameshifts kleine verschuivingen die de code helemaal veranderen.
Voorbeeld van dat laatste stel je hebt iets dat codeert voor Alanine A het triplet GCA. Als je dan de RNA code GCA-GCA-GCA hebt staat dat voor AAA, maar als je nu 1 base weghaalt krijg je bijvoorbeeld GCG-CAG-CAG wat staat voor AQQ heel iets anders dus.
SPOILER: code tabelOm spoilers te kunnen lezen moet je zijn ingelogd. Je moet je daarvoor eerst gratis Registreren. Ook kun je spoilers niet lezen als je een ban hebt.Verder hebben ze het over de polyA staart dat is een groepje As wat aan het einde van functionele RNAs gehangen wordt zodat ze herkent worden door de afleesmechanismen. Blijkbaar heeft dit virus die staart in zijn genoom zitten maar wordt hij bij het processen iets verkort.En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Er is nu een brazilliaanse variant toegevoegd die heel erg afwijkt. Begin van een zuidamerikaanse variant?
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
weet niet of het hier thuishoort?, het is een reeks van twitter berichten van een USA dokter.
https://twitter.com/davidasinclair/status/1238972083864027137
https://twitter.com/davidasinclair/status/1238972083864027137
Interessant.quote:
weet niet of het hier thuishoort?, het is een reeks van twitter berichten van een USA dokter.
https://twitter.com/davidasinclair/status/1238972083864027137
Die nieuwe mutatie op >20.quote:
Er is nu een brazilliaanse variant toegevoegd die heel erg afwijkt. Begin van een zuidamerikaanse variant?
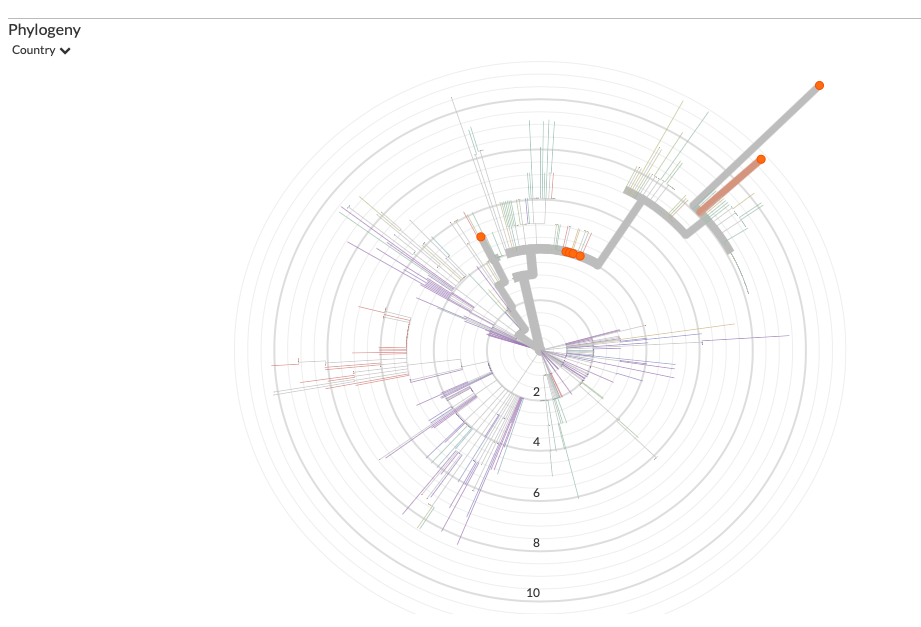
Kan het ook zijn dat het in de US is begonnen en dat zij daardoor zo'n specifieke afwijkende tak hebben? Het is toch vreemd dat ze met zo'n afstand mutaties vinden? Dan missen er toch "generaties"?
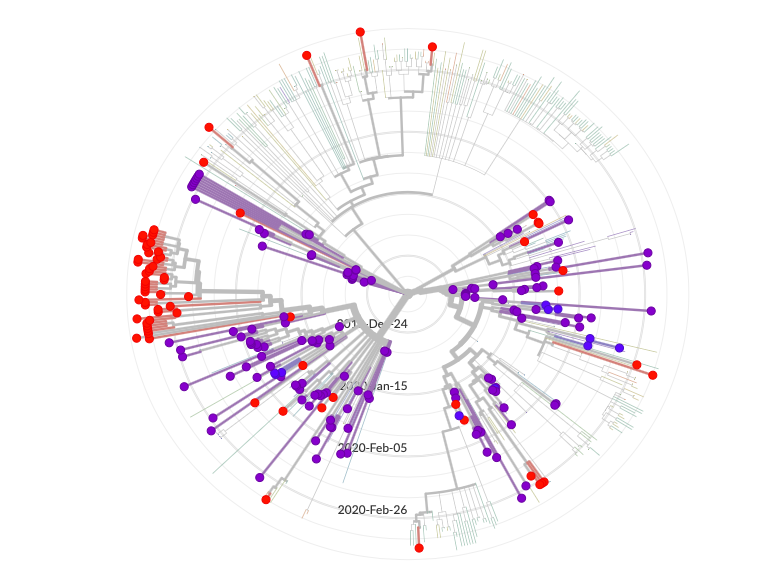
Zeker in tijd ziet de US (rood) eruit als een redelijke geïsoleerde vertakking.
https://nextstrain.org/nc(...)n,USA,China&l=radial
Er kunnen ook veel mutaties in één keer bij komen of een grote deletie oid.quote:Op dinsdag 17 maart 2020 09:25 schreef _I het volgende:
[..]
Die nieuwe mutatie op >20.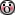
Kan het ook zijn dat het in de US is begonnen en dat zij daardoor zo'n specifieke afwijkende tak hebben? Het is toch vreemd dat ze met zo'n afstand mutaties vinden? Dan missen er toch "generaties"?
[ afbeelding ]
Die amerikaanse heeft 12 mutaties ten opzichte van zijn voorganger, dat kun je zien als je op de stipjes hovert.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
AH, dacht dat het altijd per kleine stapjes ging.quote:
[..]
Er kunnen ook veel mutaties in één keer bij komen of een grote deletie oid.
Heeft het DNA van de gastheer daar nog iets mee van doen? Of komt de codering volledig vanuit het RNA van het virus zelf?
En las dat als twee virussen 1 cel binnendringen er combinaties kunnen ontstaan tot een nieuw virus. Zou dat hier kunnen spelen?
(Begrijp het helemaal als je er moe van wordt.
Ik weet niet hoe corona het doet maar RNA virussen (corona is een RNA virus met een enkele streng RNA, dus niet dubbel zoals bij DNA maar 1 helft ) hebben vaak RNA polymerases aan boord, deze kunnen RNA moleculen kopieren. Er zit bij dit geen kopieren geen of weinig "proofreading" waardoor er sneller fouten in de kopie ontstaan.quote:
[..]
AH, dacht dat het altijd per kleine stapjes ging.
Heeft het DNA van de gastheer daar nog iets mee van doen? Of komt de codering volledig vanuit het RNA van het virus zelf?
En las dat als twee virussen 1 cel binnendringen er combinaties kunnen ontstaan tot een nieuw virus. Zou dat hier kunnen spelen?
(Begrijp het helemaal als je er moe van wordt.)
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Om de zaken nog wat lastiger te maken heeft corona ook eiwitten die weer in door zichzelf in kleine stukjes geknipt wordt en die kleine stukken samen vormen de RNA polymerase
https://www.nwo.nl/onderz(...)ecten/i/87/2487.htmlquote:The coronavirus replicase/transcriptase is an extremely complex ~7000 amino acid protein that is autoproteolytically cleaved into 16 nonstructural proteins (nsp1-16), which are assumed to be the active enzymes in viral RNA synthesis. Among these are key enzymes for viral RNA synthesis, like the RNA-dependent RNA polymerase (RdRp; nsp12) and helicase (nsp13) that are found in other +RNA viruses, but also many poorly defined proteins that appear to be unique for coronaviruses and their closest relatives.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna

Thx.quote:
[..]
Ik weet niet hoe corona het doet maar RNA virussen (corona is een RNA virus met een enkele streng RNA, dus niet dubbel zoals bij DNA maar 1 helft ) hebben vaak RNA polymerases aan boord, deze kunnen RNA moleculen kopieren. Er zit bij dit geen kopieren geen of weinig "proofreading" waardoor er sneller fouten in de kopie ontstaan.
En andersom? Zou het kunnen zorgen voor een mutatie bij de mens?
* _I start google op.
Voor zover ik weet zit er in corona geen reverse transcriptase een eiwit dat RNA omzet naar DNA, daardoor is het moeilijker om voor corona zijn genetisch materiaal aan de gastheer door te geven.quote:Op dinsdag 17 maart 2020 09:53 schreef _I het volgende:
[..]
Thx.
En andersom? Zou het kunnen zorgen voor een mutatie bij de mens?
* _I start google op.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Dat scheelt een hoop speurwerk.quote:
[..]
Voor zover ik weet zit er in corona geen reverse transcriptase een eiwit dat RNA omzet naar DNA, daardoor is het moeilijker om voor corona zijn genetisch materiaal aan de gastheer door te geven.
voor de achtergrond.quote:
DNA <-> RNA -> eiwit
dat is hoe het zit dat peiltje terug van RNA naar DNA is niet heel veel voorkomend, en op je DNA sla je dingen op en RNA gebruik je vooral. DNA kun je zien een instructieboek met post-its en notities waardoor niet het hele boek op elk moment leesbaar is. RNA zijn receptjes op servetjes geschreven die als het gerecht klaar is de prullebak in gaan, en soms ook gebruikt worden om iets op te vegen of een vliegtuigje van te vouwen.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Mooie metafoor.quote:
[..]
voor de achtergrond.
DNA <-> RNA -> eiwit
dat is hoe het zit dat peiltje terug van RNA naar DNA is niet heel veel voorkomend, en op je DNA sla je dingen op en RNA gebruik je vooral. DNA kun je zien een instructieboek met post-its en notities waardoor niet het hele boek op elk moment leesbaar is. RNA zijn receptjes op servetjes geschreven die als het gerecht klaar is de prullebak in gaan, en soms ook gebruikt worden om iets op te vegen of een vliegtuigje van te vouwen.
Fascinerend om het complete samenspel te zien tussen zeer complexe zaken, die allemaal opgebouwd zijn uit dezelfde 4 basis elementen.
Niet alleen door dit virus, ook door een genetisch onderzoek wat ik begin dit jaar heb ondergaan, veel gelezen over dit onderwerp.
Ben niet gelovig, maar het gedeelte wat in mijn hersenen is gemaakt voor dat doel, is volledig gewijd aan de wetenschap en alle fundamentele elementen die daar aan ten grondslag liggen.
Linkquote:Mutations can reveal how the coronavirus moves—but they’re easy to overinterpret
By Kai KupferschmidtMar. 9, 2020 , 1:00 PM
Immediately after Christian Drosten published a genetic sequence of the novel coronavirus online on 28 February, he took to Twitter to issue a warning. As the virus has raced around the world, more than 350 genome sequences have been shared on the online platform GISAID. They hold clues to how the new virus, named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is spreading and evolving. But because the sequences represent a tiny fraction of cases and show few telltale differences, they are easy to overinterpret, as Drosten realized.
A virologist at the Charité University Hospital in Berlin, he had sequenced the virus from a German patient infected with COVID-19 in Italy. The genome looked similar to that of a virus found in a patient in Munich, the capital of Bavaria, more than 1 month earlier; both shared three mutations not seen in early sequences from China. Drosten realized this could give rise to the idea that the Italian outbreak was “seeded” by the one in Bavaria, which state public health officials said had been quashed by tracing and quarantining all contacts of the 14 confirmed cases. But he thought it was just as likely that a Chinese variant carrying the three mutations had taken independent routes to both countries. The newly sequenced genome “is not sufficient to claim a link between Munich and Italy,” Drosten tweeted.
His warning went unheeded. A few days later, Trevor Bedford of the Fred Hutchinson Cancer Research Center, who analyzes the stream of viral genomes and discusses them in Twitter threads, wrote that the pattern suggested the outbreak in Bavaria had not been contained after all, and appeared to have led to the Italian outbreak. The analysis spread widely. Technology Review asserted that “the Munich event could be linked to a decent part of the overall European outbreak” and Twitter users called on Germany to apologize. (This Science correspondent retweeted Bedford’s thread as well.)
Virologist Eeva Broberg of the European Centre for Disease Prevention and Control agrees with Drosten that there are more plausible scenarios for how the disease reached northern Italy than an undetected spread from Bavaria. Other scientists say Bedford jumped the gun as well. “I have to kick his butt a bit for this,” says Richard Neher, a computational biologist at the University of Basel who works with Bedford. “It’s a cautionary tale,” says Andrew Rambaut, a molecular evolutionary biologist at the University of Edinburgh. “There is no way you can make that claim just from the phylogeny alone.” Bedford later clarified he believed it was equally plausible there had been two separate introductions from China. “I think I should have been more careful with that Twitter thread,” he says.
It was a case study in the power and pitfalls of real-time analysis of viral genomes. “This is an incredibly important disease. We need to understand how it is moving,” says Bette Korber, a biologist at Los Alamos National Laboratory who is also studying the genome of SARS-CoV-2. “With very limited evolution during the outbreak, [these researchers] are doing what they can and they are making suggestions, which I think at this point should be taken as suggestions.”
As the outbreak unfolds, we expect to see more and more diversity. … And then it will become easier and easier to actually put things together.
The sequence data were most informative early on, says Kristian Andersen, a computational biologist at Scripps Research. The very first sequence, in early January, answered the most basic question: What pathogen is causing the disease? The ones that followed were almost identical, strongly suggesting there was a single introduction from an animal into the human population. If the virus had jumped the species barrier multiple times, scientists would see more variety among the first human cases.
Now, more diversity is emerging. Like all viruses, SARS-CoV-2 evolves over time through random mutations, only some of which are caught and corrected by the virus’s error correction machinery. Over the length of its 30,000-base-pair genome, SARS-CoV-2 accumulates an average of about one to two mutations per month, Rambaut says. “It’s about two to four times slower than the flu,” he says. Using these little changes, researchers can draw up phylogenetic trees, much like family trees. They can also make connections between different cases of COVID-19 and gauge whether there might be undetected spread of the virus.
For instance, when researchers sequenced the second virus genome in Washington—from a teenager diagnosed with COVID-19 on 27 February—it looked like a direct descendant of the first genome, a case found 6 weeks earlier, that had acquired three further mutations. Bedford tweeted that he considered it “highly unlikely” that the two genomes came from separate introductions. “I believe we are facing an already substantial outbreak in Washington State that was not detected until now,” he wrote. That analysis turned out to be correct: Washington has now reported more than 100 cases and 15 deaths and additional genomes from other patients have bolstered the link. In this case, Bedford’s hypothesis was much stronger because the two patients both came from Snohomish County, Rambaut says: “It’s very unlikely that this highly related virus would travel to exactly the same town in Washington,” he says.
Few other firm conclusions about the virus’s spread have emerged, in part because the wealth of genomes is still a tiny sample of the more than 100,000 cases worldwide. Although China accounts for 80% of all COVID-19 cases, only one-third of the published genomes are from China—and very few of them are from later cases. And because it’s early in the outbreak, most genomes are still very similar, which makes it hard to draw conclusions. “We just have this handful of mutations, which makes these groupings so ambiguous,” Neher says. “As the outbreak unfolds, we expect to see more and more diversity and more clearly distinct lineages,” he says. “And then it will become easier and easier to actually put things together.”
Scientists will also be scouring the genomic diversity for mutations that might change how dangerous the pathogen is or how fast it spreads. There, too, caution is warranted. A paper published by Lu Jian of Peking University and colleagues on 3 March in the journal National Science Review analyzed 103 virus genomes and argued that they fell into one of two distinct types, named S and L, distinguished by two mutations. Because 70% of sequenced SARS-CoV-2 genomes belong to L, the newer type, the authors concluded that virus has evolved to become more aggressive and to spread faster.
But they lack evidence, Rambaut says. “What they’ve done is basically seen these two branches and said, ‘That one is bigger, [so that virus] must be more virulent or more transmissible,’” he says. However, just because a virus is exported and leads to a large outbreak elsewhere does not mean it is behaving differently: “One of these lineages is going to be bigger than the other just by chance.” Some researchers have called for the paper to be retracted. “The claims made in it are clearly unfounded and risk spreading dangerous misinformation at a crucial time in the outbreak,” four scientists at the University of Glasgow wrote in a response published on www.virological.org. (In a response, Lu wrote the four had misunderstood his study.)
Most genomic changes don’t alter the virus’s behavior, Drosten says. The only way to confirm that a mutation has an effect is to study it in cell cultures or animal models and show, for instance, that it has become better at entering cells or transmitting, he says. And if the virus does change in an important way, it could go either way, making it more or less dangerous. In 2018, Drosten’s group published a paper showing that early in the SARS outbreak of 2002–03, that virus lost a small chunk of its genome, 29 base pairs in one gene. Adding those base pairs back in the lab made the virus much better at replicating in several cell culture models.
It might seem strange that a mutation that weakens the virus would become established, but that can happen when it has just entered the human population and isn’t competing with strains lacking the mutation, Drosten says. “Sadly, this new virus doesn’t have that deletion,” he adds.
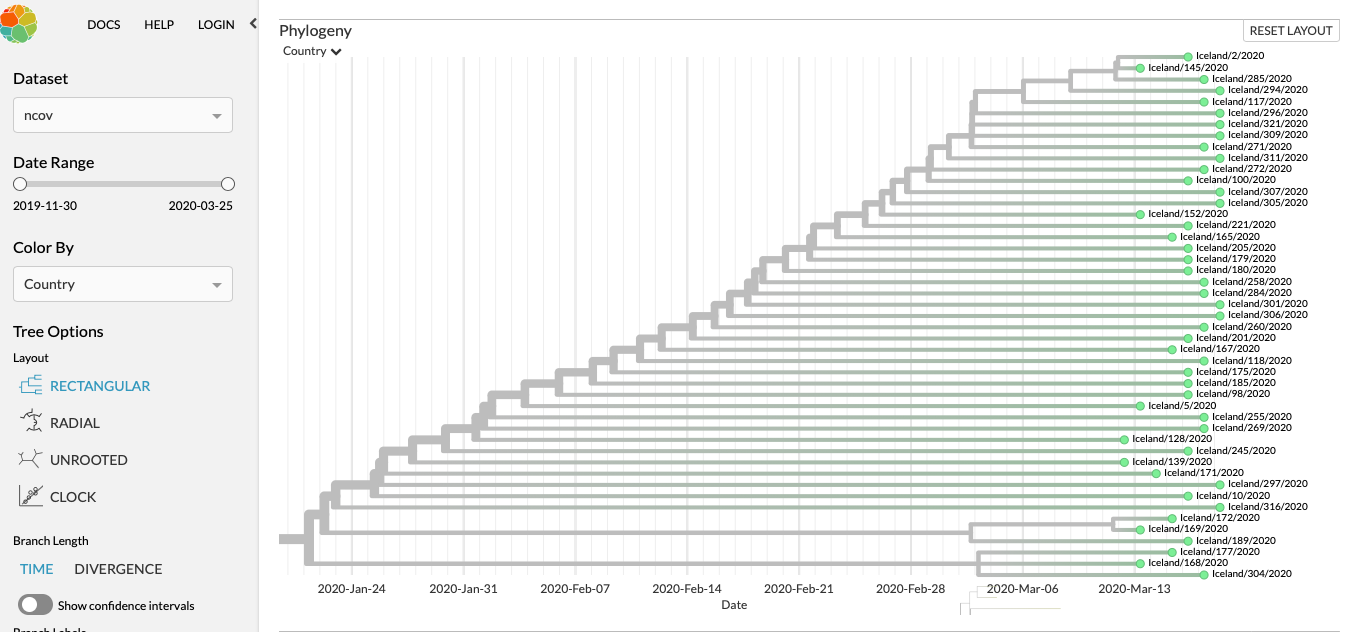
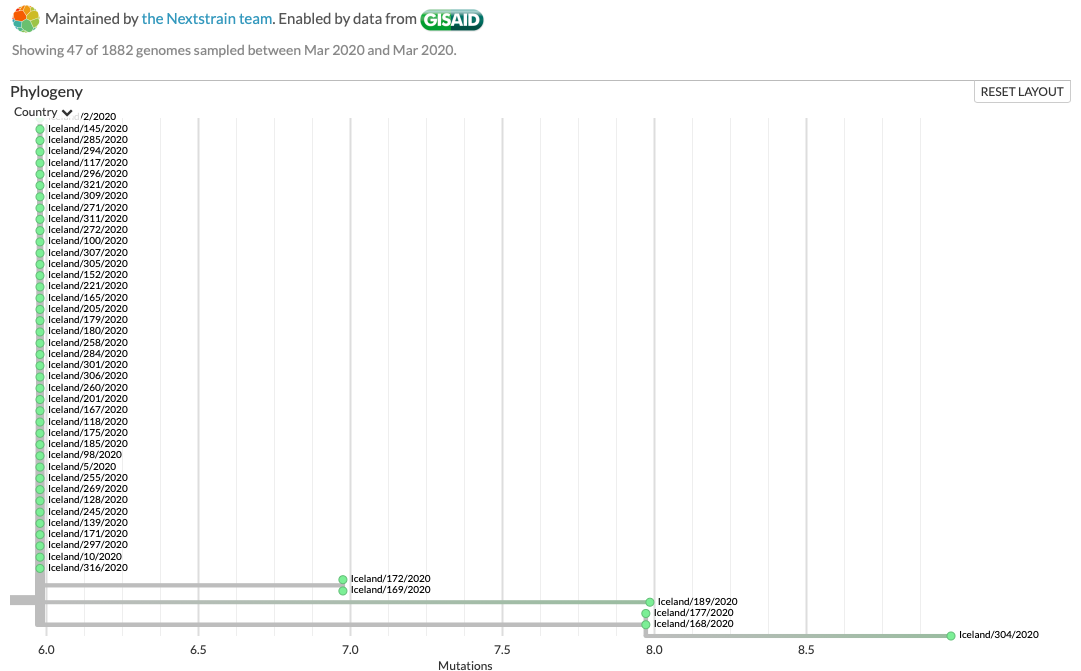
Van de 711 genomen komen er 166 uit NL.
Het filter staat alleen aan voor NL en voor ORF8 site 84; De befaamde L en S strain.
De L zou "agressiever" zijn dan de S; als dit zo is, had ik liever iets meer -Geel- gezien.
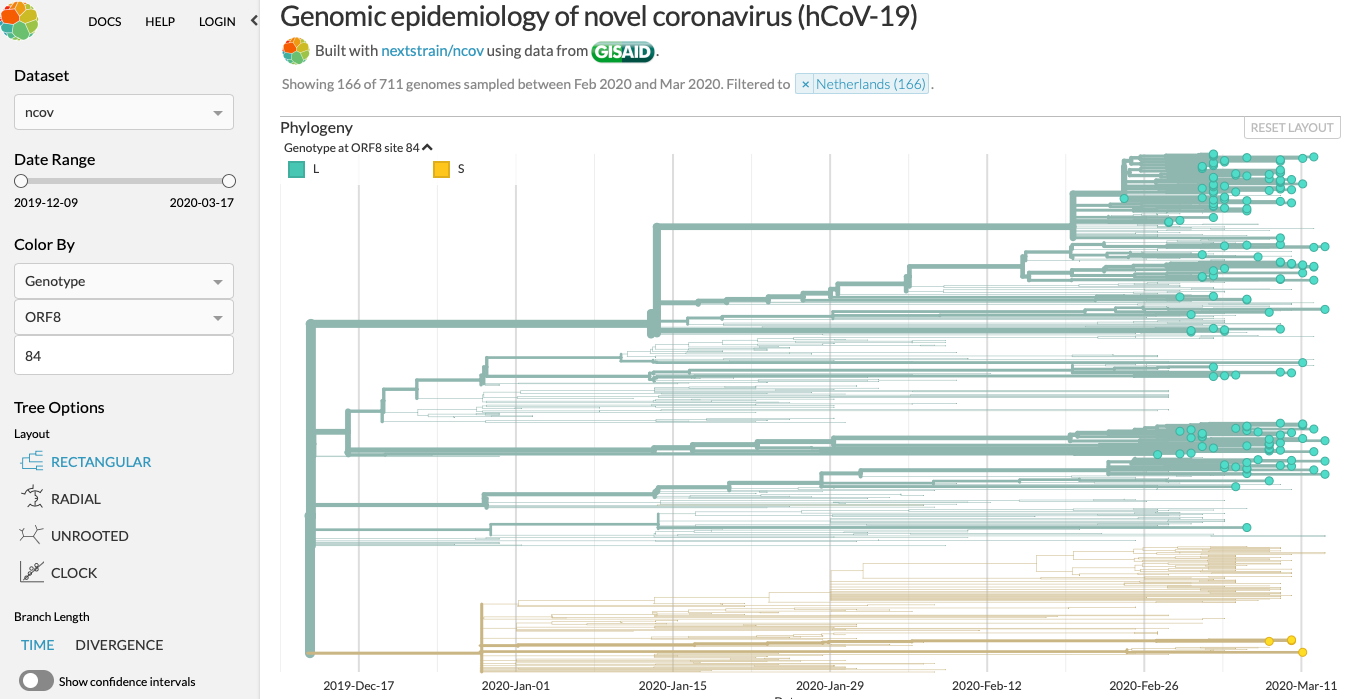
wat is een springer?quote:
De US heeft een "springer". Zo.
[ afbeelding ]
Geen wetenschappelijke term, hence de aanhalingstekens. Het beschrijft slechts de sprong in mutaties van het rode balletje helemaal rechts.quote:
Ook maar hier tegen ondersneeuwing
quote:Ik zit in de trein de nature van deze week te lezen, en in een artikel daarin laten ze zien dan deze corona bindt aan het humane ACE2 eiwit en bijvoorbeeld niet aan die van muizen, muizen kunnen dus niet geïnfecteerd worden.
Maar ze testen daar ook ACE2 van civet katten, vleermuizen en varkens. En die binden allemaal aan deze corona, nou zou ik me niet zo druk maken om de vleermuizen en civetkatten. Maar ik vraag me wel af of onze varkenspopulatie gechecked wordt... En of dat een extra bron kan zien zijn\worden?
Bron: Zhou et.al. 2020 nature
In dezelfde nature staat ook dat tests om makaken laten zien dat die slechts milde effecten hebben, ben benieuwd of ze erachter komen wat de mens zo heftig doet reageren.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
twitter:nextstrain twitterde op donderdag 19-03-2020 om 17:15:57 Thanks to #opendata sharing by Zhejiang University School of Medicine & @GISAID, we've updated https://t.co/SDPCOcsnMX with two new #COVID19 #SARSCOV2 #hCoV19 sequences from Hangzhou, China.
As shown in big blue circles below, they fall in two difference places on the tree. https://t.co/o80otMghk2 reageer retweet
 Thanks to
Thanks to 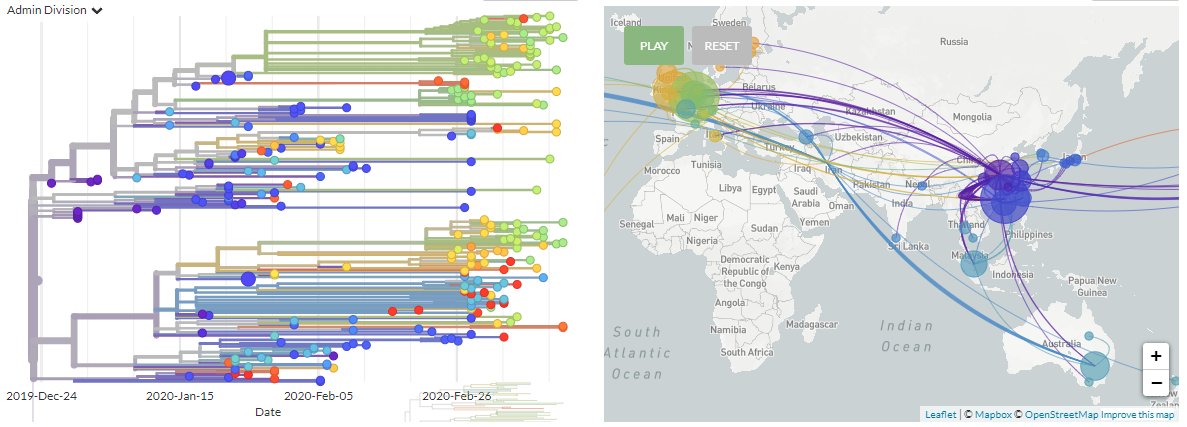
Enorme veranderingen je ziet nu amerikaanse maar ook een belgisch/franse tak.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna

US en UK
FR en BE
NL
================
Zonder enige verdere kennis, de twee mutaties die hierboven ook al genoemd zijn op -S- en -ORF8-
Mutatie op -S-; waarbij een gedeelte van onze besmettingen in de ene categorie vallen en deels in de andere (let op dat ene verdwaalde gele bolletje tussen al het groen
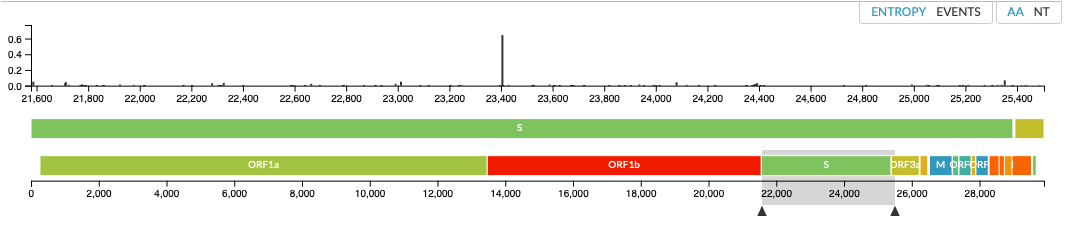
En de befaamde mutatie op ORF8; de -L- en -S- variant
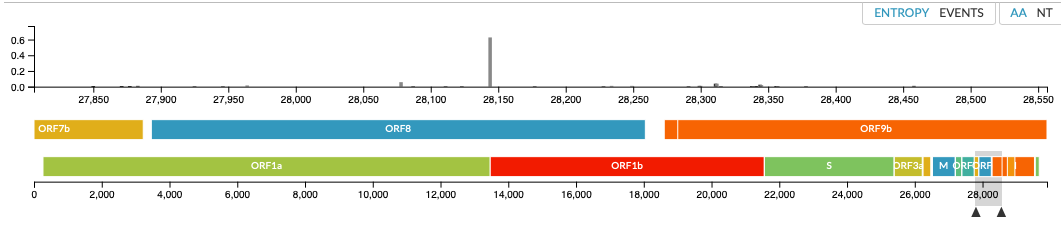
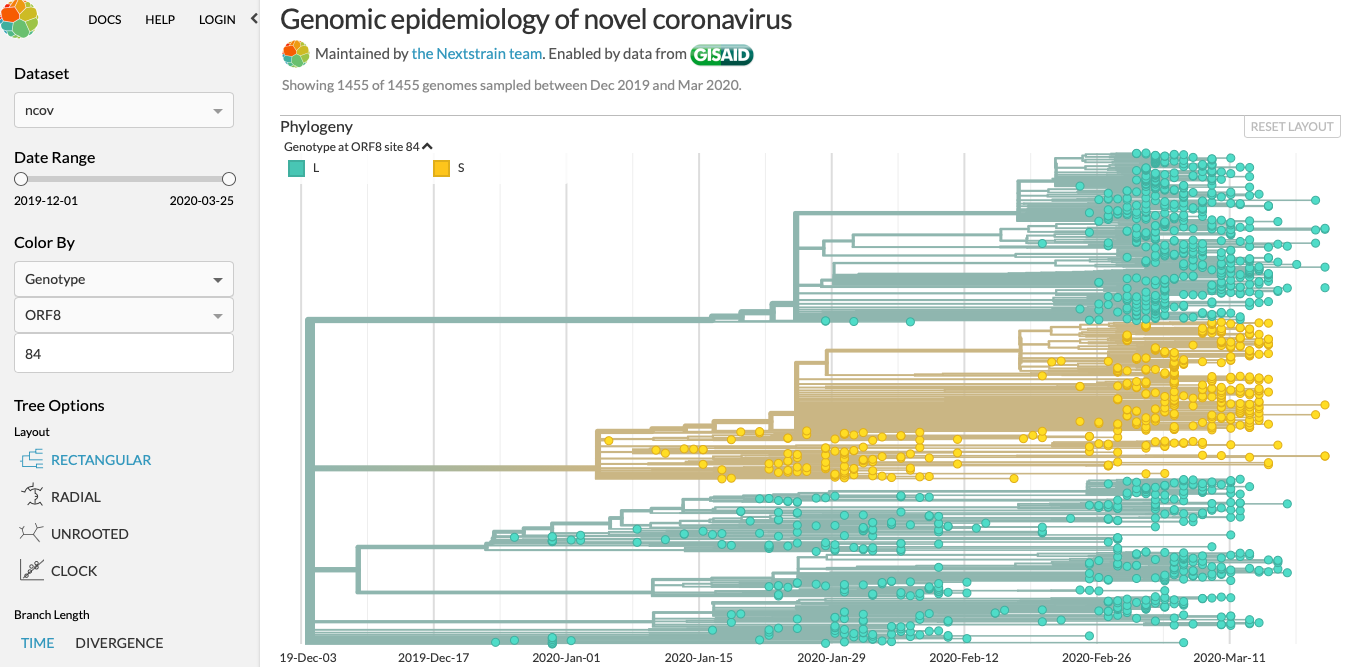
Waarbij de -S- vooral in de USA heerst. (Rood is USA)
Zeer interessant heren. 
Ik denk meer dat je als nieuwsposter een geile egocentrische narcist moet zijn, die een flinke stijve krijgt van alle berichten die ie van zijn eigen hand ziet op de FP, zo! ©yvonne
Beste nieuwsbericht ooit op de FOK!frontpage!
Beste nieuwsbericht ooit op de FOK!frontpage!
Genetica schept duidelijkheid: Covid-19 is gewoon SARS
RD - Dr. Peter Borger
Na een paar weken coronacrisis lijken de nieuwsmedia het over één ding eens: er is een nieuw coronavirus opgedoken. Maar is dat wel zo?
Het coronavirus dat momenteel de ronde doet en Covid-19 verspreidt, is door een internationaal consortium van virusonderzoekers SARS-CoV2 gedoopt. Waarom met de toevoeging ”2”? Dat hebben ze gedaan omdat het virus sprekend lijkt op het coronavirus dat in 2003 SARS verbreidde, en als SARS-CoV te boek staat. Eigenlijk zou het virus uit 2003 nu SARS-CoV1 kunnen heten.
De symptomen die SARS-CoV1 en SARS-CoV2 voortbrengen, de ziektebeelden van SARS en Covid-19, zijn nauwelijks van elkaar te onderscheiden. Een longarts zou niet kunnen zeggen of een patiënt is geïnfecteerd met SARS-CoV1 of met SARS-CoV2. Dat kunnen wetenschappers alleen vaststellen door het op moleculair niveau te testen.
Het genetische materiaal van SARS-CoV2 komt namelijk voor 96,11 procent overeen met een SARS-virusstam die RaTG13 wordt genoemd. Het SARS-CoV2-virus heeft dezelfde genen als het SARS-CoV1 virus van 2003; het dringt op precies dezelfde manier door in de menselijke cel, via de zogeheten ACE2-receptor.
In zeventien jaar tijd is het virus gelukkig een beetje veranderd. Door mutaties worden virussen door de jaren heen steeds zwakker. Dat komt doordat dergelijke RNA-virussen zeer snel muteren. Elke keer als hun genen worden gekopieerd, zullen er willekeurige kopieerfoutjes optreden. Soms worden er kleine stukjes genetisch materiaal toegevoegd of gaan kleine stukjes verloren. Het is zeer waarschijnlijk, dat op deze manier uit SARS-CoV1 de variant SARS-CoV2 is ontstaan. Of dit in een vleermuis, in een gordeldier of in een laboratorium in Wuhan gebeurde, zullen we nooit weten.
Besmettelijker
Een constante ophoping van mutaties maakt RNA-virussen minder dodelijk, maar wel besmettelijker. Virusvarianten die mensen echt ziek maken of doden, zullen immers uitsterven. Zo was ook de H1N1-varkensgriep het gevaarlijkst tijdens de eerste uitbraak in de zomer van 2009, maar in de winter van 2011 was het al afgezwakt tot niet meer dan een typische seizoensgriep. SARS-CoV1 begon in 2003 als een nieuw en zeer gevaarlijk coronavirus. In de loop van zeventien jaar zwakte het af en muteerde tot SARS-CoV2.
Het verschil tussen beide virussen zit voornamelijk in vier kleine mutaties in het zogeheten S-gen. Dat codeert voor het zogeheten spike-protein, waarmee het virus de cellen binnendringt. Daardoor is het virus besmettelijker dan SARS-CoV uit 2003. We hebben dus zeer waarschijnlijk te maken met hetzelfde virus, hoewel de wereldgezondheidsorganisatie WHO met de naam ”Covid-19” in plaats van ”SARS” hierover enige verwarring schept.
Verstoppen
Als het om hetzelfde coronavirus gaat, waar heeft het zich dan al die tijd verstopt? Virussen overleven op twee verschillende manieren. Allereerst in dieren. Vleermuizen en vogels worden vaak –en terecht– aangehaald als bronnen van virussen. Maar ook andere dieren zijn als gastheer geschikt. Veelal worden deze dierlijke gastheren zelf niet ziek omdat ze een ander, wellicht sterker immuunsysteem hebben dan mensen.
De andere manier om te overleven, is in biomedische laboratoria, waar wetenschappers virussen bestuderen om er medicatie en vaccins tegen te ontwikkelen. Zo overleeft het pokkenvirus, dat officieel geldt als uitgeroeid, momenteel in een tank met vloeibare stikstof in twee militaire laboratoria in de USA en in Rusland.
Het is bekend dat er niet met het coronavirus werd geknoeid: het is geen product van doelgerichte genetische manipulatie. Dat zou een deskundige meteen herkennen.
Het genetische materiaal van SARS-CoV2 is eenduidig: dit virus is op een natuurlijke wijze ontstaan.
Chimpansee
De grote genetische overeenkomst –96,11 procent– tussen SARS-CoV1 en SARS-CoV2 duidt er sterk op dat we te maken hebben met hetzelfde virus. Als echter dezelfde methode zou worden gebruikt als darwinistische evolutiebiologen hanteren wanneer ze de genen van mensen en chimpanzees met elkaar vergelijken, zijn de genomen van beide virussen zelfs voor meer dan 99 procent gelijk.
Alle argumenten samen maken slechts één conclusie mogelijk: SARS-CoV1 en SARS-CoV2 zijn twee varianten van hetzelfde virus. Dat maakt het extra wrang dat het WHO samen met de regeringen de SARS-onderzoeksprogramma’s hebben stopgezet. Medicijnen en vaccins tegen SARS waren nu zeer waarschijnlijk ook effectief gebleken tegen Covid-19.
De auteur werkte twintig jaar als moleculair bioloog aan luchtwegziekten en is auteur van ”Terug naar de Oorsprong” (2009) en ”Darwin Revisited” (2018).
RD - Dr. Peter Borger
Na een paar weken coronacrisis lijken de nieuwsmedia het over één ding eens: er is een nieuw coronavirus opgedoken. Maar is dat wel zo?
Het coronavirus dat momenteel de ronde doet en Covid-19 verspreidt, is door een internationaal consortium van virusonderzoekers SARS-CoV2 gedoopt. Waarom met de toevoeging ”2”? Dat hebben ze gedaan omdat het virus sprekend lijkt op het coronavirus dat in 2003 SARS verbreidde, en als SARS-CoV te boek staat. Eigenlijk zou het virus uit 2003 nu SARS-CoV1 kunnen heten.
De symptomen die SARS-CoV1 en SARS-CoV2 voortbrengen, de ziektebeelden van SARS en Covid-19, zijn nauwelijks van elkaar te onderscheiden. Een longarts zou niet kunnen zeggen of een patiënt is geïnfecteerd met SARS-CoV1 of met SARS-CoV2. Dat kunnen wetenschappers alleen vaststellen door het op moleculair niveau te testen.
Het genetische materiaal van SARS-CoV2 komt namelijk voor 96,11 procent overeen met een SARS-virusstam die RaTG13 wordt genoemd. Het SARS-CoV2-virus heeft dezelfde genen als het SARS-CoV1 virus van 2003; het dringt op precies dezelfde manier door in de menselijke cel, via de zogeheten ACE2-receptor.
In zeventien jaar tijd is het virus gelukkig een beetje veranderd. Door mutaties worden virussen door de jaren heen steeds zwakker. Dat komt doordat dergelijke RNA-virussen zeer snel muteren. Elke keer als hun genen worden gekopieerd, zullen er willekeurige kopieerfoutjes optreden. Soms worden er kleine stukjes genetisch materiaal toegevoegd of gaan kleine stukjes verloren. Het is zeer waarschijnlijk, dat op deze manier uit SARS-CoV1 de variant SARS-CoV2 is ontstaan. Of dit in een vleermuis, in een gordeldier of in een laboratorium in Wuhan gebeurde, zullen we nooit weten.
Besmettelijker
Een constante ophoping van mutaties maakt RNA-virussen minder dodelijk, maar wel besmettelijker. Virusvarianten die mensen echt ziek maken of doden, zullen immers uitsterven. Zo was ook de H1N1-varkensgriep het gevaarlijkst tijdens de eerste uitbraak in de zomer van 2009, maar in de winter van 2011 was het al afgezwakt tot niet meer dan een typische seizoensgriep. SARS-CoV1 begon in 2003 als een nieuw en zeer gevaarlijk coronavirus. In de loop van zeventien jaar zwakte het af en muteerde tot SARS-CoV2.
Het verschil tussen beide virussen zit voornamelijk in vier kleine mutaties in het zogeheten S-gen. Dat codeert voor het zogeheten spike-protein, waarmee het virus de cellen binnendringt. Daardoor is het virus besmettelijker dan SARS-CoV uit 2003. We hebben dus zeer waarschijnlijk te maken met hetzelfde virus, hoewel de wereldgezondheidsorganisatie WHO met de naam ”Covid-19” in plaats van ”SARS” hierover enige verwarring schept.
Verstoppen
Als het om hetzelfde coronavirus gaat, waar heeft het zich dan al die tijd verstopt? Virussen overleven op twee verschillende manieren. Allereerst in dieren. Vleermuizen en vogels worden vaak –en terecht– aangehaald als bronnen van virussen. Maar ook andere dieren zijn als gastheer geschikt. Veelal worden deze dierlijke gastheren zelf niet ziek omdat ze een ander, wellicht sterker immuunsysteem hebben dan mensen.
De andere manier om te overleven, is in biomedische laboratoria, waar wetenschappers virussen bestuderen om er medicatie en vaccins tegen te ontwikkelen. Zo overleeft het pokkenvirus, dat officieel geldt als uitgeroeid, momenteel in een tank met vloeibare stikstof in twee militaire laboratoria in de USA en in Rusland.
Het is bekend dat er niet met het coronavirus werd geknoeid: het is geen product van doelgerichte genetische manipulatie. Dat zou een deskundige meteen herkennen.
Het genetische materiaal van SARS-CoV2 is eenduidig: dit virus is op een natuurlijke wijze ontstaan.
Chimpansee
De grote genetische overeenkomst –96,11 procent– tussen SARS-CoV1 en SARS-CoV2 duidt er sterk op dat we te maken hebben met hetzelfde virus. Als echter dezelfde methode zou worden gebruikt als darwinistische evolutiebiologen hanteren wanneer ze de genen van mensen en chimpanzees met elkaar vergelijken, zijn de genomen van beide virussen zelfs voor meer dan 99 procent gelijk.
Alle argumenten samen maken slechts één conclusie mogelijk: SARS-CoV1 en SARS-CoV2 zijn twee varianten van hetzelfde virus. Dat maakt het extra wrang dat het WHO samen met de regeringen de SARS-onderzoeksprogramma’s hebben stopgezet. Medicijnen en vaccins tegen SARS waren nu zeer waarschijnlijk ook effectief gebleken tegen Covid-19.
De auteur werkte twintig jaar als moleculair bioloog aan luchtwegziekten en is auteur van ”Terug naar de Oorsprong” (2009) en ”Darwin Revisited” (2018).
Stomme vraag wellicht maar kan je dan niet het eerdere onderzoek afmaken en op korte termijn dat vaccin toepassen als het 99% hetzelfde is?quote:
Genetica schept duidelijkheid: Covid-19 is gewoon SARS
RD - Dr. Peter Borger
Na een paar weken coronacrisis lijken de nieuwsmedia het over één ding eens: er is een nieuw coronavirus opgedoken. Maar is dat wel zo?
Het coronavirus dat momenteel de ronde doet en Covid-19 verspreidt, is door een internationaal consortium van virusonderzoekers SARS-CoV2 gedoopt. Waarom met de toevoeging ”2”? Dat hebben ze gedaan omdat het virus sprekend lijkt op het coronavirus dat in 2003 SARS verbreidde, en als SARS-CoV te boek staat. Eigenlijk zou het virus uit 2003 nu SARS-CoV1 kunnen heten.
De symptomen die SARS-CoV1 en SARS-CoV2 voortbrengen, de ziektebeelden van SARS en Covid-19, zijn nauwelijks van elkaar te onderscheiden. Een longarts zou niet kunnen zeggen of een patiënt is geïnfecteerd met SARS-CoV1 of met SARS-CoV2. Dat kunnen wetenschappers alleen vaststellen door het op moleculair niveau te testen.
Het genetische materiaal van SARS-CoV2 komt namelijk voor 96,11 procent overeen met een SARS-virusstam die RaTG13 wordt genoemd. Het SARS-CoV2-virus heeft dezelfde genen als het SARS-CoV1 virus van 2003; het dringt op precies dezelfde manier door in de menselijke cel, via de zogeheten ACE2-receptor.
In zeventien jaar tijd is het virus gelukkig een beetje veranderd. Door mutaties worden virussen door de jaren heen steeds zwakker. Dat komt doordat dergelijke RNA-virussen zeer snel muteren. Elke keer als hun genen worden gekopieerd, zullen er willekeurige kopieerfoutjes optreden. Soms worden er kleine stukjes genetisch materiaal toegevoegd of gaan kleine stukjes verloren. Het is zeer waarschijnlijk, dat op deze manier uit SARS-CoV1 de variant SARS-CoV2 is ontstaan. Of dit in een vleermuis, in een gordeldier of in een laboratorium in Wuhan gebeurde, zullen we nooit weten.
Besmettelijker
Een constante ophoping van mutaties maakt RNA-virussen minder dodelijk, maar wel besmettelijker. Virusvarianten die mensen echt ziek maken of doden, zullen immers uitsterven. Zo was ook de H1N1-varkensgriep het gevaarlijkst tijdens de eerste uitbraak in de zomer van 2009, maar in de winter van 2011 was het al afgezwakt tot niet meer dan een typische seizoensgriep. SARS-CoV1 begon in 2003 als een nieuw en zeer gevaarlijk coronavirus. In de loop van zeventien jaar zwakte het af en muteerde tot SARS-CoV2.
Het verschil tussen beide virussen zit voornamelijk in vier kleine mutaties in het zogeheten S-gen. Dat codeert voor het zogeheten spike-protein, waarmee het virus de cellen binnendringt. Daardoor is het virus besmettelijker dan SARS-CoV uit 2003. We hebben dus zeer waarschijnlijk te maken met hetzelfde virus, hoewel de wereldgezondheidsorganisatie WHO met de naam ”Covid-19” in plaats van ”SARS” hierover enige verwarring schept.
Verstoppen
Als het om hetzelfde coronavirus gaat, waar heeft het zich dan al die tijd verstopt? Virussen overleven op twee verschillende manieren. Allereerst in dieren. Vleermuizen en vogels worden vaak –en terecht– aangehaald als bronnen van virussen. Maar ook andere dieren zijn als gastheer geschikt. Veelal worden deze dierlijke gastheren zelf niet ziek omdat ze een ander, wellicht sterker immuunsysteem hebben dan mensen.
De andere manier om te overleven, is in biomedische laboratoria, waar wetenschappers virussen bestuderen om er medicatie en vaccins tegen te ontwikkelen. Zo overleeft het pokkenvirus, dat officieel geldt als uitgeroeid, momenteel in een tank met vloeibare stikstof in twee militaire laboratoria in de USA en in Rusland.
Het is bekend dat er niet met het coronavirus werd geknoeid: het is geen product van doelgerichte genetische manipulatie. Dat zou een deskundige meteen herkennen.
Het genetische materiaal van SARS-CoV2 is eenduidig: dit virus is op een natuurlijke wijze ontstaan.
Chimpansee
De grote genetische overeenkomst –96,11 procent– tussen SARS-CoV1 en SARS-CoV2 duidt er sterk op dat we te maken hebben met hetzelfde virus. Als echter dezelfde methode zou worden gebruikt als darwinistische evolutiebiologen hanteren wanneer ze de genen van mensen en chimpanzees met elkaar vergelijken, zijn de genomen van beide virussen zelfs voor meer dan 99 procent gelijk.
Alle argumenten samen maken slechts één conclusie mogelijk: SARS-CoV1 en SARS-CoV2 zijn twee varianten van hetzelfde virus. Dat maakt het extra wrang dat het WHO samen met de regeringen de SARS-onderzoeksprogramma’s hebben stopgezet. Medicijnen en vaccins tegen SARS waren nu zeer waarschijnlijk ook effectief gebleken tegen Covid-19.
De auteur werkte twintig jaar als moleculair bioloog aan luchtwegziekten en is auteur van ”Terug naar de Oorsprong” (2009) en ”Darwin Revisited” (2018).
Daar zullen ze vast wel mee bezig zijn, neem ik aan. Als men er geld in ziet.quote:
[..]
Stomme vraag wellicht maar kan je dan niet het 2009 onderzoek afmaken en op korte termijn dat vaccin toepassen?
Heb er verder niet heel veel kijk op.
Dat is ook de reden dat o.a. het erasmusmc maar ook vele andere groepen al vrij snel wat zaken hadden liggen. Echter medicatie echt naar de mensen krijgen is een stuk moeilijker en duurt zelfs met omzeilen van regels over het algemeen minimaal maanden tot jaren.quote:Op donderdag 26 maart 2020 16:26 schreef Cesare-Borgia het volgende:
[..]
Stomme vraag wellicht maar kan je dan niet het eerdere onderzoek afmaken en op korte termijn dat vaccin toepassen als het 99% hetzelfde is?
Je wilt niet na de corona uitbraak allemaal doden op je geweten hebben dankzij ondeugelijke medicatie. Dat hebben we van o.a. softenon wel geleerd.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
Is er al een uberstrain ontstaan?
"This is your life and it's ending one minute at a time." - Tyler Durden
"Sand is overrated. It's just tiny, little rocks." - Joel
"Sand is overrated. It's just tiny, little rocks." - Joel

Definieer uberstrain en nee.quote:
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
laatste dagen is hij wel van de complot theorie.....zou het witte huis hem even opgebeld hebben ?quote:
woensdag 28 oktober 2020 15:54 schreef Kyran het volgende:[/b]
Even screenshot gemaakt van dit moment. Predator en sorry zeggen
Even screenshot gemaakt van dit moment. Predator en sorry zeggen
Interessant topic en interessant artikel. Wat een enorm gemiste kans dat de onderzoeksprogramma's tegen SARS zijn stopgezet door de WHO. Nu zijn de enorme economische en sociale gevolgen van dit slechte beleid te zien. Ik hoop dat hier echt lering uit getrokken gaat worden.quote:
Genetica schept duidelijkheid: Covid-19 is gewoon SARS
RD - Dr. Peter Borger
Na een paar weken coronacrisis lijken de nieuwsmedia het over één ding eens: er is een nieuw coronavirus opgedoken. Maar is dat wel zo?
Het coronavirus dat momenteel de ronde doet en Covid-19 verspreidt, is door een internationaal consortium van virusonderzoekers SARS-CoV2 gedoopt. Waarom met de toevoeging ”2”? Dat hebben ze gedaan omdat het virus sprekend lijkt op het coronavirus dat in 2003 SARS verbreidde, en als SARS-CoV te boek staat. Eigenlijk zou het virus uit 2003 nu SARS-CoV1 kunnen heten.
De symptomen die SARS-CoV1 en SARS-CoV2 voortbrengen, de ziektebeelden van SARS en Covid-19, zijn nauwelijks van elkaar te onderscheiden. Een longarts zou niet kunnen zeggen of een patiënt is geïnfecteerd met SARS-CoV1 of met SARS-CoV2. Dat kunnen wetenschappers alleen vaststellen door het op moleculair niveau te testen.
Het genetische materiaal van SARS-CoV2 komt namelijk voor 96,11 procent overeen met een SARS-virusstam die RaTG13 wordt genoemd. Het SARS-CoV2-virus heeft dezelfde genen als het SARS-CoV1 virus van 2003; het dringt op precies dezelfde manier door in de menselijke cel, via de zogeheten ACE2-receptor.
In zeventien jaar tijd is het virus gelukkig een beetje veranderd. Door mutaties worden virussen door de jaren heen steeds zwakker. Dat komt doordat dergelijke RNA-virussen zeer snel muteren. Elke keer als hun genen worden gekopieerd, zullen er willekeurige kopieerfoutjes optreden. Soms worden er kleine stukjes genetisch materiaal toegevoegd of gaan kleine stukjes verloren. Het is zeer waarschijnlijk, dat op deze manier uit SARS-CoV1 de variant SARS-CoV2 is ontstaan. Of dit in een vleermuis, in een gordeldier of in een laboratorium in Wuhan gebeurde, zullen we nooit weten.
Besmettelijker
Een constante ophoping van mutaties maakt RNA-virussen minder dodelijk, maar wel besmettelijker. Virusvarianten die mensen echt ziek maken of doden, zullen immers uitsterven. Zo was ook de H1N1-varkensgriep het gevaarlijkst tijdens de eerste uitbraak in de zomer van 2009, maar in de winter van 2011 was het al afgezwakt tot niet meer dan een typische seizoensgriep. SARS-CoV1 begon in 2003 als een nieuw en zeer gevaarlijk coronavirus. In de loop van zeventien jaar zwakte het af en muteerde tot SARS-CoV2.
Het verschil tussen beide virussen zit voornamelijk in vier kleine mutaties in het zogeheten S-gen. Dat codeert voor het zogeheten spike-protein, waarmee het virus de cellen binnendringt. Daardoor is het virus besmettelijker dan SARS-CoV uit 2003. We hebben dus zeer waarschijnlijk te maken met hetzelfde virus, hoewel de wereldgezondheidsorganisatie WHO met de naam ”Covid-19” in plaats van ”SARS” hierover enige verwarring schept.
Verstoppen
Als het om hetzelfde coronavirus gaat, waar heeft het zich dan al die tijd verstopt? Virussen overleven op twee verschillende manieren. Allereerst in dieren. Vleermuizen en vogels worden vaak –en terecht– aangehaald als bronnen van virussen. Maar ook andere dieren zijn als gastheer geschikt. Veelal worden deze dierlijke gastheren zelf niet ziek omdat ze een ander, wellicht sterker immuunsysteem hebben dan mensen.
De andere manier om te overleven, is in biomedische laboratoria, waar wetenschappers virussen bestuderen om er medicatie en vaccins tegen te ontwikkelen. Zo overleeft het pokkenvirus, dat officieel geldt als uitgeroeid, momenteel in een tank met vloeibare stikstof in twee militaire laboratoria in de USA en in Rusland.
Het is bekend dat er niet met het coronavirus werd geknoeid: het is geen product van doelgerichte genetische manipulatie. Dat zou een deskundige meteen herkennen.
Het genetische materiaal van SARS-CoV2 is eenduidig: dit virus is op een natuurlijke wijze ontstaan.
Chimpansee
De grote genetische overeenkomst –96,11 procent– tussen SARS-CoV1 en SARS-CoV2 duidt er sterk op dat we te maken hebben met hetzelfde virus. Als echter dezelfde methode zou worden gebruikt als darwinistische evolutiebiologen hanteren wanneer ze de genen van mensen en chimpanzees met elkaar vergelijken, zijn de genomen van beide virussen zelfs voor meer dan 99 procent gelijk.
Alle argumenten samen maken slechts één conclusie mogelijk: SARS-CoV1 en SARS-CoV2 zijn twee varianten van hetzelfde virus. Dat maakt het extra wrang dat het WHO samen met de regeringen de SARS-onderzoeksprogramma’s hebben stopgezet. Medicijnen en vaccins tegen SARS waren nu zeer waarschijnlijk ook effectief gebleken tegen Covid-19.
De auteur werkte twintig jaar als moleculair bioloog aan luchtwegziekten en is auteur van ”Terug naar de Oorsprong” (2009) en ”Darwin Revisited” (2018).
Article 1 Universal Declaration of Human Rights
'All human beings are born free and equal in dignity and rights. They are endowed with reason and conscience and should act towards one another in a spirit of brotherhood.'
'All human beings are born free and equal in dignity and rights. They are endowed with reason and conscience and should act towards one another in a spirit of brotherhood.'
Crosspost
quote:
Voor de geïnteresseerden er staat een erg goed overzichts artikel in nature van 3 mei
https://www.nature.com/articles/d41586-020-01315-7
Wat ik opvallend vond en nog niet wist is de proofreading capaciteit van het virus en dat het virus blijkbaar recombineert.
En mochten we vallen dan is het omhoog. - Krang (uit: Pantani)
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
My favourite music is the music I haven't yet heard - John Cage
Water: ijskoud de hardste - Gehenna
|
|
| Forum Opties | |
|---|---|
| Forumhop: | |
| Hop naar: | |